Urednik sekcije: prof. dr. sc. Milan Vrkljan, dr. med.
Prijevod: Božidar Perić, dr. med.
HEMOKROMATOZA
Hemokromatoza je poremećaj pohrane željeza koji dovodi do povećane apsorpcije željeza iz crijeva te odlaganja željeza i oštećenja mnogih tkiva. Klasična klinička konstelacija hemokromatoze je bolesnik, koji se prezentira brončanom kožom, jetrenom bolesti, šećernom bolesti, artropatijom, poremećajima provođenja srčanog impulsa i hipogonadizmom. Postoje dva glavna uzroka: nasljedni (zbog nasljednih mutantnih HFE gena) i sekundarno opterećenje željezom (uobičajeno zbog neučinkovite eritropoeze, poput talasemije ili sideroblastične anemije). HFE kodira bjelančevinu uključenu u prepoznavanje staničnog željeza i u regulaciji apsorpcije željeza iz crijeva. Mutacije HFE gena su vrlo česte u populaciji Sjeverne Europe (1 od 10 su nositelji). Heterozigoti su asimptomatski; homozigoti pokazuju prodornost bolest ~30%. Postoji progresivno prekomjerno nakupljanje željeza, s kliničkim manifestacijama koje se javljaju iza 30–40 godina, tipično ranije kod muškaraca, nego u žena. Alkoholna bolest jetre i kronični ekscesivni unos željeza mogu biti povezani s umjerenim porastom jetrenog željeza i povišenih zaliha željeza u tijelu.
Klinička slika
Rani simptomi su slabost, malaksalost, mršavljenje, brončana pigmentacija ili tamnjenje kože, bol u trbuhu i gubitak libida. Hepatomegalija nastaje u 95% bolesnika, katkad uz uredne testove jetrene funkcije. Neliječena, jetrena bolest napreduje do ciroze i dalje do hepatocelularnog karcinoma u ~30% bolesnika s cirozom. Ostali znakovi su brončana pigmentacija kože, šećerna bolest (65% bolesnika), artropatija (25–59%), srčane aritmije i srčana dekompenzacija (15%) te hipogonadotropni hipogonadizam. Šećerna bolest je češća kod bolesnika s pozitivnom obiteljskom anamnezom šećerne bolesti, a hipogonadizam može biti izolirana rana manifestacija. Tipični znakovi portalne hipertenzije i dekompenzirane jetrene ciroze mogu se javiti kasnije u kliničkom tijeku. Adrenalna insuficijencija, hipotireoza i hipoparatireoidizam rijetko se javljaju.
Dijagnoza
Povišene su vrijednosti serumskog željeza, postotka zasićenja transferina i razine serumskog feritina. U inače zdrave osobe saturacija serumskog transferina natašte >50% je patološka i ukazuje na homozigotnost za hemokromatozu. U većine neliječenih bolesnika s hemokromatozom razina serumskog feritina je također u velikoj mjeri povišena. Bilo da je postotak zasićenja transferina ili razina serumskog feritina nenormalna, treba izvršiti genetske testove na hemokromatozu. Sve krvne srodnike u prvom koljenu oboljelih od hemokromatoze treba testirati na HFE mutacije C282Y i H63D. Biopsija jetre može biti potrebna radi procjene mogućeg nastanka ciroze ili kvantifikacije željeza u tkivima. Algoritam za procjenjivanje bolesnika s mogućom hemokromatozom prikazuje Sl. 179-1. Smrt neliječenih bolesnika posljedica je srčane dekompenzacije (30%), ciroze (25%) i hepatocelularnog karcinoma (30%); potonje se može razviti unatoč primjerenom odstranjivanju Fe.
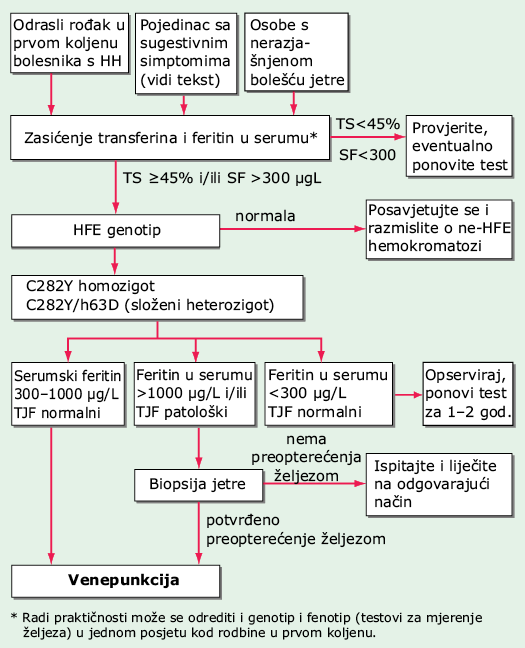
SLIKA 179-1 Algoritam za probir hemokromatoze povezane s HFE. HH = hereditarna (nasljedna) hemokromatoza, homozigotna osoba (C282Y +/+); LFT = testovi jetrene funkcije (engl. liver function tests); SF = koncentracija feritina u serumu; TS = zasićenje transferina. (Iz EJ Eijkelkamp i suradnici: Can J Gastroenterol 14: 2, 2000; uz dopuštenje.)
LIJEČENJE HEMOKROMATOZA
Liječenje se sastoji od uklanjanja suviška tjelesnog željeza, obično povremenim venepunkcijama te potpornih terapijskih mjera za oštećene organe. Kako 1 jedinica krvi sadrži ∼250 mg Fe, a mora se ukloniti ≥25 g Fe, venepunkcija se vrši jednom tjedno tijekom 1–2 godine. Rjeđe se venepunkcije nakon toga rade u cilju održanja serumskog Fe na razini 9–18 μmol/L (50–100 μg/dl). Kelirajuća sredstva kao što je deferoksamin (daje se supkutano preko portabilne pumpe) uklanjaju 10–20 mg željeza na dan, dio onog mobiliziranog tjednom venepunkcijom. Međutim, terapija kelatima indicirana je kada je venepunkcija neprimjerena, npr. u slučaju anemije ili hipoproteinemije. Konzumiranje alkohola treba zabraniti. Terminalni stadij jetrene bolesti može zahtijevati potrebu za transplantacijskim liječenjem.
PORFIRIJE
Porfirije su nasljedni poremećaji biosinteze hema. Svaki od 9 poremećaja uzrokuje jedinstvenu sliku prekomjerne proizvodnje, nakupljanja i izlučivanja intermedijarnih produkata sinteze hema. Ovi se poremećaji dijele na hepatične i eritropoetske, ovisno o primarnom sijelu prekomjerne proizvodnje i nakupljanja preteča porfirina ili porfirina. Glavne manifestacije hepatičnih porfirija su neurološke (neuropatska bol u trbuhu, neuropatija i duševni poremećaji), dok je za eritropoetske porfirije karakteristična fotosenzitivnost kože. Laboratorijske pretrage su potrebne za potvrdu ili isključenje različitih vrsta porfirija. Međutim, za konačnu dijagnozu treba dokazati manjak specifičnog enzima ili genski defekt. Ovdje su opisana samo tri najčešća tipa porfirije.
AKUTNA INTERMITENTNA PORFIRIJA
To je autosomno dominantan poremećaj koji se različito iskazuje, a uzrokovan je djelomičnim (50%) deficitom hidroksimetilbilan sintaze. Ima prevalenciju 1–3 na 100.000, ali je nešto češći u određenim dijelovima svijeta (Sjeverna Švedska, Velika Britanija). Manifestacije su abdominalna bol poput kolika, povraćanje, konstipacija, urin boje crnog vina te neurološki i psihijatrijski poremećaji. Akutni napadi su rijetki prije puberteta, a mogu trajati danima ili mjesecima. Fotosenzitivnost se ne javlja. Kliničke i biokemijske manifestacije mogu pospješiti barbiturati, antikonvulzivi, estrogeni, oralni kontraceptivi, alkohol ili niskokalorična prehrana. Dijagnoza se postavlja dokazivanjem porasta porfobilinogena (PBG) i γ-aminolevulinske kiseline (ALA) u mokraći za vrijeme akutnog napada. Genetsko testiranje, ukoliko je dostupno, trebalo bi se izvršiti radi potvrde dijagnoze.
LIJEČENJE AKUTNA INTERMITENTNA PORFIRIJA
Čim dođe do napada, treba što prije dati 3–4 mg hema u obliku hem arginata, hem albumina ili hematina, u infuziji svaki od uzastopna 4 dana. Hem djeluje inhibirajući ALA sintazu, obuzdavajući pritom proizvodnju ALA i PBG. Akutni se napadi mogu učinkovito suzbiti davanjem IV glukoze brzinom do 20 g/h ili parenteralnom prehranom, ako duže razdoblje nije moguće provoditi hranjenje na usta. Tijekom akutnih napada radi bolova u trbuhu mogu biti potrebni opioidni analgetici, a fenotiazini su korisni za mučninu, povraćanje, tjeskobu i nemir. Liječenje između napada uključuje uzimanje odgovarajuće hrane, izbjegavanje lijekova za koje se zna da mogu pogoršati bolest i brzo liječenje drugih interkurentnih bolesti ili infekcija. Presađivanje jetre se pokazalo učinkovitim u probranih bolesnika, a u tijeku su istraživanja zamjene gena.
PORPHYRIA CUTANEA TARDA
Ovo je najčešća porfirija (2–4 na 100.000), za koju je karakteristična fotosenzitivnost kože i, obično, jetrena bolest. Nastaje zbog manjka (naslijeđenog ili stečenog) jetrene uroporfirinogen dekarboksilaze. Fotosenzitivnost uzrokuje pigmentaciju lica, pojačanu krhkost kože, eritem te vezikularne i ulcerozne lezije, koje u pravilu zahvaćaju lice, čelo i podlaktice. Neurološke manifestacije nisu zamijećene. Doprinoseći čimbenici su višak alkohola, željeza i estrogena. Bolesnici s jetrenom bolešću su pod većim rizikom ciroze i hepatocelularnog karcinoma. U plazmi i mokraći su povišeni uroporfirin i 7-karboksilat porfirin.
LIJEČENJE PORPHYRIA CUTANEA TARDA
U prvu terapijsku liniju ubrajamo izbjegavanje faktora koji pospješuju bolest, uključujući apstinenciju od alkohola, estrogena, pripravaka željeza i drugih lijekova koji mogu izazvati pogoršanje. Potpun terapijski odgovor može se gotovo uvijek postići ponavljanjem venepunkcije (svakih 1–2 tjedna), sve dok se ne smanji hepatično željezo. Bolesnicima koji nisu u stanju podnijeti ili ne reagiraju na venepunkciju mogu se dati klorokin ili hidroksiklorokin u malim dozama (npr. 125 mg klorokin fosfata dvaput tjedno), da se poboljša izlučivanje porfirina.
ERITROPOETIČNA PROTOPORFIRIJA
Eritropoetična porfirija je autosomno dominantan poremećaj zbog djelomičnog deficita ferokelataze, posljednjeg enzima na putu biosinteze hema. Prevalencija iznosi 1 na 100.000. Porfirini (primarno protoporfirin IX) iz eritrocita koštane moždine i plazme, odlažu se u kožu dovodeći do fotosenzitivnosti kože. Fotosenzitivnost kože obično počinje u djetinjstvu. Kožne se manifestacije razlikuju od onih kod drugih porfirija, utoliko što su vezikularne lezije rijetke. Unutar nekoliko minuta od izlaganja sunčevoj svjetlosti može se razviti crvenilo, edem, pečenje i svrbež i nalikuju angioedemu. Simptomi se mogu činiti neproporcionalni vidljivim kožnim lezijama. Kronične kožne promjene mogu uključiti lihenifikaciju, kožaste pseudovezikule, izbrazdale usne i promjene na noktima. Jetrena funkcija obično je uredna iako se mogu javiti jetrena bolest i žučni kamenci. Razina protoporfirina je povišena u koštanoj moždini, cirkulirajućim eritrocitima, plazmi, žuči i stolici; protoporfirini u eritrocitima su u većem omjeru slobodni, u manjoj mjeri vezani za cink, za razliku od ostalih porfirija i hematoloških poremećaja. Razina porfirina u mokraći je uredna. Dijagnozu potvrđuje otkrivanje mutacije na genu za enzim ferokelatazu.
LIJEČENJE ERITROPOETIČNA PROTOPORFIRIJA
Neophodno je izbjegavati sunce. Peroralni β-karoten (120–180 mg/d) poboljšava podnošenje sunčeve svjetlosti kod mnogih bolesnika. Dozu treba prilagoditi tako da razina serumskog karotena bude između 10 i 15 μmol/L (600–800 μg/dl). Kolestiramin ili aktivni ugljen mogu olakšati izlučivanje protoporfirina stolicom. Plazmafereza ili intravenska primjena hema mogu biti od koristi.
WILSONOVA BOLEST
Wilsonova bolest je rijetki nasljedni poremećaj metabolizma bakra koji dovodi do toksičnog nakupljanja bakra u jetri, mozgu i drugim organima. Osobe s Wilsonovom bolesti imaju mutacije na genu ATP7B, koji kodira membranom vezanu adenozin trifosfatazu (ATP-azu), koja služi za prijenos bakra. Deficit ove bjelančevine otežava izlučivanje bakra u bilijarni sustav, kao i ugradnju bakra u ceruloplazmin, dovodeći do njegove ubrzane razgradnje.
Klinička slika
Kliničke manifestacije se tipično javljaju u srednjoj i kasnijoj tinejdžerskoj dobi, no mogu se javiti i kasnije. Bolest jetre se može očitovati kao hepatitis, ciroza ili jetrena dekompenzacija. Kod drugih su bolesnika neurološki ili psihijatrijski poremećaji prvi klinički znak i uvijek ih prati pojava Kayser-Fleischerova prstena (depoziti bakra u rožnici). Distonija, poremećaj koordinacije ili tremor mogu biti prisutni, a disartrija i disfagija su česta pojava. Isto tako, mogući su i autonomni poremećaji. Mikrohematurija je uobičajena. U oko 5% bolesnika prva manifestacija može biti primarna ili sekundarna amenoreja ili ponavljani spontani pobačaj.
Dijagnoza
Razina serumskog ceruloplazmina obično je niska, iako može biti uredna u do oko 10% bolesnika. Razina bakra u mokraći je povišena. “Zlatni standard” za postavljanje dijagnoze je povišena razina bakra u uzorku jetre dobivenom biopsijom. Genetsko testiranje može biti potvrdno, ali poremećaj može biti posljedica velikog broja različitih mutacija.
LIJEČENJE WILSONOVA BOLEST
Hepatitis ili cirozu bez dekompenzacije treba liječiti cink-acetatom (50 mg elementarnog cinka PO 3× dnevno). Cink pokazuje učinkovitost sprječavajući apsorpciju bakra iz probavnog sustava i indukciju metalotioneina, koji izolira bakar iz netoksičnog kompleksa. Bolesnicima s jetrenom dekompenzacijom preporučuje se kelator trienten (500 mg PO 2× dnevno) plus cink (u razmaku od barem 1 sata radi izbjegavanja keliranja cinka u intestinalnom lumenu), premda u slučaju teške jetrene dekompenzacije treba razmotriti transplantaciju jetre. Za inicijalnu neurološku terapiju preporučuje se trientin i cink kroz 8 tjedana, a zatim se liječenje nastavlja samo cinkom. Tetratiomolibdat je alternativna terapijska mogućnost raspoloživa u budućnosti. Penicilamin više nije lijek prvog izbora. Liječenje cinkom ne zahtijeva kontrolu koncentracije zbog toksičnosti, a terapijski odgovor se prati određivanjem bakra u 24-satnoj mokraći. Trientin može izazvati supresiju koštane moždine i proteinuriju. S kelirajućom terapijom za praćenje terapijskog učinka, primarno se preporuča mjerenje razine slobodnog bakra u serumu (podešavanje ukupni serumski bakar za ceruloplazminski bakar), u odnosu na određivanje bakra u urinu. Terapija se mora provoditi doživotno.
Opširnije vidi u Powell LW: Hemochromatosis, Pogl. 428, str. 2514; Desnick RJ, Balwani M: The Porphyrias, Pogl. 430, str. 2521; Brewer GJ: Wilson’s Disease, Pogl. 429, str. 2519, u HPIM-19.