AKUTNA MIJELOIČNA LEUKEMIJA (AML)
AML je klonalna maligna bolest mijeloidnih prekursorskih stanica koštane srži u kojoj dolazi do nakupljanja slabo diferenciranih stanica u koštanoj srži i cirkulaciji.
Simptomi bolesti nastaju zbog nedostatka zrelih stanica koje koštana srž normalno proizvodi, uključujući granulocite (podložnost infekcijama) i trombocite (sklonost krvarenju). Osim toga, ako se u cirkulaciji nalazi veliki broj nezrelih malignih mijeloblasta, oni mogu infiltrirati organe i u rijetkim slučajevima dovesti do disfunkcije. Postoje različiti morfološki podtipovi (Tbl. 65-1) koji se u velikoj mjeri klinički preklapaju. Vrijedi napomenuti da bolesnici s akutnom promijelocitnom leukemijom (APL) (FAB M3) imaju sklonost krvarenju i diseminiranoj intravaskularnoj koagulaciji, osobito tijekom indukcijske kemoterapije, zbog oslobađanja prokoagulanata iz citoplazmatskih granula.
TABLICA 65-1 SUSTAVI ZA KLASIFIKACIJU AML
|
|
KLASIFIKACIJA SVJETSKE ZDRAVSTVENE ORGANIZACIJE a
|
|
AML s recidivirajućim genetskim poremećajima
|
|
AML s t(8;21)(q22;q22); RUNX1-RUNX1T1b
|
|
AML s inv(16)(p13.1;1q22) ili t(16;16)(p13.1;q22); CBFB-MYH11b
|
|
Akutna promijelocitna leukemija s t(15;17)(q22;q12); PML-RARAb
|
|
AML s t(9;11)(p22;q23); MLLT3-MLL
|
|
AML s t(6;9)(p23;q34); DEK-NUP214
|
|
AML s inv(3)(q21q26.2) ili t(3;3)(q21;q26.2); RPN1-EVI1
|
|
AML (megakarioblastična) s t(1;22)(p13;q13); RBM15-MKL1
|
|
Privremeno stanje: AML s mutiranim NPM1
|
|
Privremeno stanje: AML s mutiranim CEBPA
|
|
AML s promjenama povezanim s mijelodisplazijom
|
|
Mijeloične neoplazme povezane s terapijom
|
|
Nespecificirane AML
|
|
AML s minimalnom diferencijacijom
|
|
AML bez sazrijevanja
|
|
AML sa sazrijevanjem
|
|
Akutna mijelomonocitna leukemija
|
|
Akutna monoblastična i monocitna leukemija
|
|
Akutna eritroidna leukemija
|
|
Akutna megakarioblastična leukemija
|
|
Akutna bazofilna leukemija
|
|
Akutna panmijeloza s mijelofibrozom
|
|
Mijeloični sarkom
|
|
Mijeloične proliferacije povezane s Downovim sindromom
|
|
Prolazna abnormalna mijelopoeza
|
|
Mijeloična leukemija povezana s Downovim sindromom
|
|
Neoplazma blastičnih plazmacitoidnih dendritičkih stanica
|
|
Akutna leukemija dvosmislene loze
|
|
Akutna nediferencirana leukemija
|
|
Akutna leukemija miješanog fenotipa s t(9;22)(q34;q11,20); BCR-ABL11
|
|
Akutna leukemija miješanog fenotipa s t(v;11q23); s promjenama MLL
|
|
Akutna leukemija miješanog fenotipa, B/mijeloična, NOS
|
|
Akutna leukemija miješanog fenotipa, T/mijeloična, NOS
|
|
Privremeno stanje: Limfoblastična leukemija/limfom NK stanica
|
|
Francusko-američko-britanska (FAB) klasifikacija c
|
|
MO: Minimalno diferencirana leukemija
|
|
Ml: Mijeloblastična leukemija bez sazrijevanja
|
|
M2: Mijeloblastična leukemija sa sazrijevanjem
|
|
M3: Hipergranularna promijelocitna leukemija
|
|
M4: Mijelomonocitna leukemija
|
|
M4Eo: Varijanta: Povećan broj abnormalnih eozinofila u koštanoj srži
|
|
M5: Monocitna leukemija
|
|
M6: Eritroleukemija (DiGuglielmova bolest)
|
|
M7: Megakarioblastična leukemija
|
|
aIz SH Swerdlow i sur (ur.): World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, IARC Press, 2008.
bbDijagnoza je AML neovisno o broju blasta.
cIz JM Bennett i sur: Ann Intern Med 103:620, 1985.
Kratice: AML = akutna mijeloična leukemija.
|
Incidencija i etiologija
U SAD-u je zabilježeno oko 20.830 novih slučajeva u 2015. godini. AML čini oko 80% akutnih leukemija u odraslih. U velikoj većini slučajeva etiologija ostaje nepoznata. Kako starimo, mogu nastati mutacije u normalnim matičnim stanicama koje prenose sposobnost proliferacije i uspostavljaju tzv. klonalnu hematopoezu. U slučaju klonalne hematopoeze relativni rizik nastanka akutne leukemije je povećan, ali je apsolutni rizik i dalje vrlo nizak. Tri štetna čimbenika iz okoliša povećavaju rizik: kronična izloženost benzenu, izloženost zračenju i prethodno liječenje alkilirajućim agensima (osobito uz radioterapiju) i inhibitorima topoizomeraze II (npr. doksorubicin i etopozid). Kronična mijeloična leukemija (KML), mijelodisplazija i mijeloproliferativni sindromi mogu prijeći u AML. Neke su genetske abnormalnosti povezane s posebnim morfološkim varijantama: t(15;17) s APL, inv(16) s eozinofilnom leukemijom; druge se javljaju u obliku više tipova. Abnormalnosti kromosoma 11q23 često se nalaze u leukemijama koje nastaju nakon izloženosti inhibitorima topoizomeraze II. Delecije (brisanje) kromosoma 5 ili 7 viđaju se u leukemijama koje nastaju nakon radiokemoterapije. Ta određena genetska abnormalnost ima velik utjecaj na ishod liječenja. Ekspresija MDR1 (multirezistentna isisna pumpa za lijekove) česta je u starijih bolesnika i najavljuje lošiji ishod (nepovoljnu prognozu).
Kliničke i laboratorijske značajke
Početni simptomi akutne leukemije obično traju <3 mjeseca. Preleukemijski sindrom može biti prisutan u oko 25% slučajeva AML-a. Najčešće nalazimo anemiju, bljedilo, umor, slabost, palpitacije i dispneju u naporu. Broj leukocita (L) može biti nizak, normalan ili izrazito povišen. Cirkulirajući blasti mogu, ali ne moraju biti prisutni. Kada su L >100×109 blasta po litri može doći do leukostaze u plućima i mozgu. Česte su manje gnojne infekcije kože. Trombocitopenija dovodi do spontanih krvarenja, epistakse, petehija, krvarenja u konjunktive, iz gingiva i do pojave modrica, osobito kada je broj trombocita <20.000/μL. Gubitak apetita i mršavljenje su česta pojava, a moguća je i vrućica.
Bakterijske i gljivične infekcije su česte. Rizik je veći kada je apsolutni broj neutrofila <5000/μL. Oštećenje sluznične i kožne barijere povećava sklonost infekcijama. U slučaju teške leukopenije infekcije mogu biti klinički neupadne pa njihovo brzo prepoznavanje zahtijeva visoki stupanj kliničke sumnje.
Hepatosplenomegaliju ima oko trećine bolesnika. Leukemijski meningitis može se manifestirati glavoboljom, mučninom, konvulzijama, edemom papile i paralizom moždanih živaca.
Metabolički poremećaji mogu biti hiponatrijemija, hipokalijemija, povišeni serumski LDH, hiperuricemija i (rijetko) laktacidoza. U slučajevima s vrlo velikim brojem blasta u krvi može doći do lažne hiperkalijemije i hipoglikemije (nakon vađenja krvi tumorske stanice oslobađaju kalij i troše glukozu).
LIJEČENJE AKUTNA MIJELOIČNA LEUKEMIJA
Masa leukemijskih stanica u trenutku otkrivanja bolesti može iznositi 1011–1012 stanica. Kada ukupni broj leukemijskih stanica padne ispod ~109 one se više ne mogu otkriti u krvi ili koštanoj srži pa se čini kao da je bolesnik u potpunoj remisiji (PR). Stoga, ako leukemiju želiom iskorijeniti mora se nastaviti s primjenom agresivne terapije nakon početnog smanjenja stanične mase. Tipične faze kemoterapije su: indukcija remisije i postremisijska terapija koja traje oko 1 godinu. Slika 65-1 prikazuje terapijski algoritam.
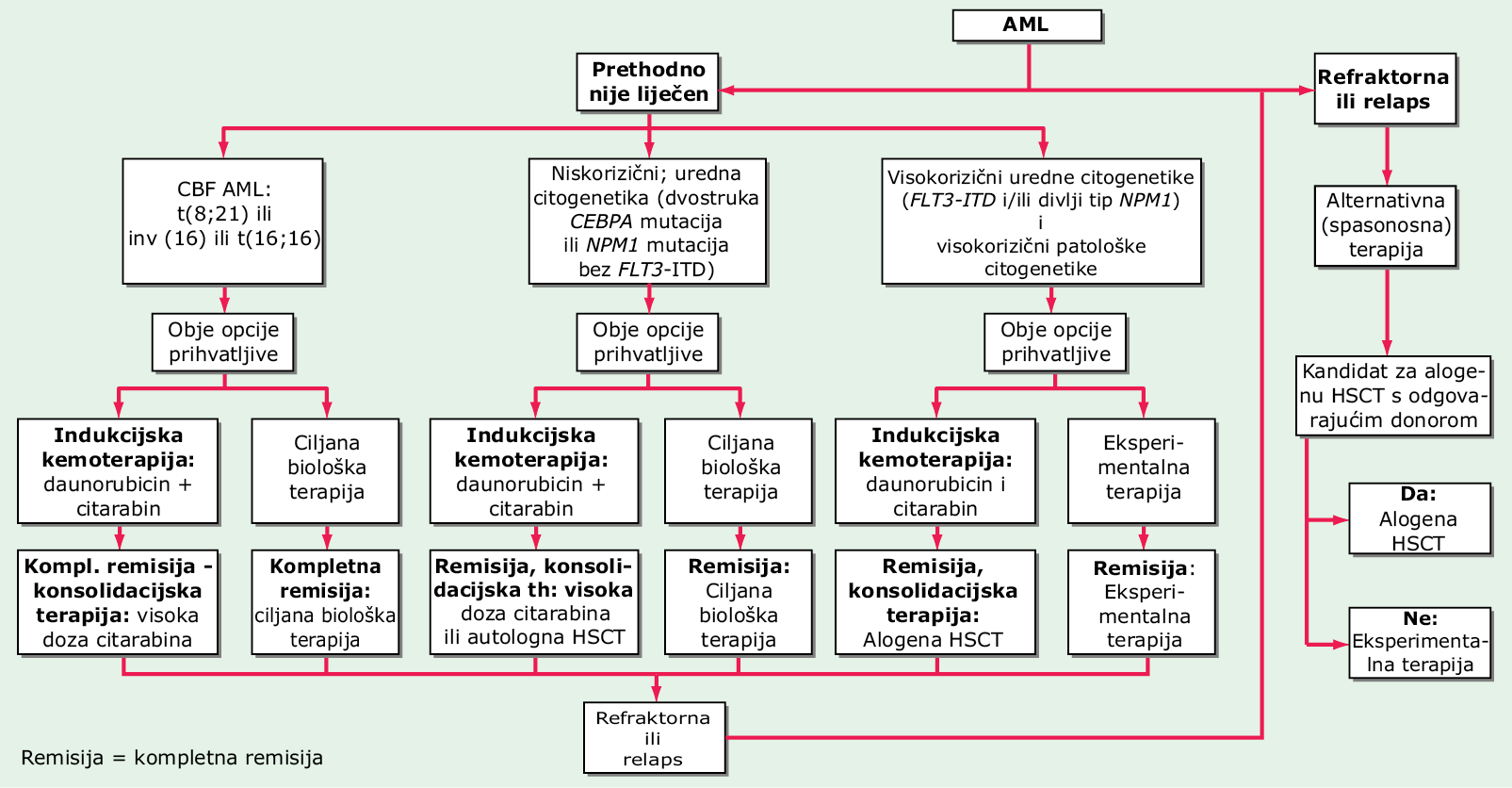
SLIKA 65-1 Terapijski dijagram za novodijagnosticiranu akutnu mijeloičnu leukemiju (AML). Za sve oblike AML-a osim akutne promijelocitne leukemije (APL) standardna terapija uključuje protokol zasnovan na 7-dnevnoj kontinuiranoj infuziji citarabina (100–200 mg/m2 dnevno) i 3-dnevnoj terapiji daunorubicinom (60–90 mg/m2 dnevno) sa ili bez drugih lijekova. Umjesto daunorubicina može se koristiti idarubicin (12–13 mg/m2 dnevno). Bolesnici koji postignu potpunu remisiju primaju postremisijsku konsolidacijsku terapiju, koja uključuje sekvencijske kure citarabina u visokim dozama, autolognu transplantaciju hematopoetskih matičnih stanica (HSCT), alogenu HSCT ili novije terapije, ovisno o riziku relapsa (npr. stratificirana terapija). Bolesnici s APL-om obično se liječe tretinoinom i arsenovim trioksidom sa ili bez antraciklinske kemoterapije, i u nekim slučajevima terapijom održavanja tretinoinom. CBF = faktor koji veže jezgru; ITD = međunarodna tandem duplikacija.
Vrlo je važna potporna terapija transfuzijama eritrocita i trombocita (od davatelja seronegativnih na CMV, ako je bolesnik kandidat za transplantaciju koštane srži) kao i agresivna prevencija, dijagnostika i liječenje infekcija. Stimulatori rasta kolonija od slabe su ili nikakve koristi. Neki ih preporučuju kod starijih bolesnika i onih s aktivnim infekcijama. Febrilnu neutropeniju treba liječiti antibioticima širokog spektra (npr. ceftazidim 1 g svakih 8 h). Ako febrilna neutropenija traje i nakon 7 dana, treba dodati amfotericin B.
Oko 60–80% bolesnika postiže inicijalnu remisiju kada se liječe protokolima s citarabinom 100–200 (mg/m2)/d u trajnoj infuziji kroz 7 dana i daunorubicinom [45 (mg/m2)/d] ili idarubicinom [12–13 (mg/m2)/d] 3 dana. Trajanje PR-a može se produžiti dodavanjem etopozida. Polovica liječenih bolesnika postiže PR nakon prvog ciklusa kemoterapije, a sljedećih 25% nakon dva ciklusa. Kod 10–30% bolesnika postiže se 5-godišnje preživljenje bez znakova bolesti, te su vjerojatno izliječeni. Bolesnici u kojih je postignuta PR i koji imaju mali rizik od recidiva [stanice sadrže t(8;21) ili inv(16)] primaju 3–4 ciklusa citarabina. Bolesnici s visokim rizikom od recidiva potencijalni su kandidati za transplantaciju alogenične koštane srži.
Terapijski odgovor nakon recidiva je kratak, a prognoza za bolesnike koji su imali recidiv je loša. Kod APL-a, arsenov trioksid plus trans-retinoična kiselina (tretinoin) inducira diferencijaciju stanica leukemije i kompletnu molekularnu remisiju. Neki bolesnici mogu imati respiratorne simptome zbog nakupljanja diferenciranih neoplastičnih granulocita u plućima. Glukokortikoidi mogu ubrzati oporavak od ovog sindroma.
Transplantacija koštane srži od identičnog blizanca ili brata ili sestre s identičnim humanim leukocitnim antigenom (HLA) je učinkovita terapija za AML. Uobičajeni protokol sastoji se od kemoterapije u visokim dozama ± ozračenja cijeloga tijela kako bi se uništila koštana srž domaćina, nakon čega se daje infuzija koštane srži davatelja. Rizik je znatan (osim u slučaju kada je koštana srž uzeta od identičnog blizanca). Komplikacije su: bolest presatka protiv domaćina, intersticijski pneumonitis, oportunističke infekcije (osobito CMV). Uspoređivanjem transplantacije i visoke doze citarabina kao postremisijske terapije nije jasno dokazana prednost nijednog od navedenih pristupa. Do 30% bolesnika, u inače terminalnom stadiju refraktorne leukemije, transplantacijom će se vjerojatno izliječiti. Rezultati su bolji kada se transplantacija izvrši tijekom remisije, a najbolji su u djece i mlađih odraslih.
KRONIČNA MIJELOIČNA LEUKEMIJA (KML)
KML je klonski malignitet koji se obično manifestira splenomegalijom i povećanim stvaranjem granulocita; tijek bolesti je u početku polagan (indolentan), ali bolest kasnije prelazi u leukemijsku fazu (blastna kriza) koja ima lošiju prognozu od de novo AML; brzina prelaska u blastnu krizu je različita; ukupno preživljenje od postavljanja dijagnoze prosječno iznosi 4 godine.
Incidencija i etiologija
U SAD-u je zabilježeno oko 14.620 novih slučajeva u 2015. godini. Više od 90% slučajeva ima recipročnu translokaciju između kromosoma 9 i 22, stvarajući Philadelphia (Ph) kromosom i produkt fuzije gena nazvan BCR-ABL. (BCR se nalazi na kromosomu 9, ABL na 22.) Kromosomska abnormalnost se javlja u svim stanicama koje potječu iz koštane srži, osim u T limfocitima. Protein koji proizvodi kimerički gen ima 210 kDa u kroničnoj fazi i 190 kDa u akutnoj blastičnoj transformaciji. Kod nekih je bolesnika kronična faza klinički nijema (asimptomatska), dok bolesnici s akutnom leukemijom imaju Ph kromosom.
Kliničke i laboratorijske značajke
Simptomi se razvijaju postupno. To su: blagi umor, malaksalost, anoreskija, mršavljenje, abdominalne tegobe u vidu nelagode i osjećaja rane sitosti zbog splenomegalije, te pretjerano znojenje. Katkada se bolesnici otkrivaju slučajno nalazom povišenog broja leukocita. Broj leukocita je obično >25.000/μL, uglavnom zbog povećanja broja granulocita i njihovih prekursora, unatrag sve do stadija mijelocita. Prevladavaju štapićasti i zreli oblici. Bazofili mogu činiti 10–15% stanica u krvi. Broj trombocita je normalan ili povećan. Anemija je česta. Razina alkalne fosfataze u neutrofilnim leukocitima je niska. Koštana srž je hipercelularna s hiperplazijom granulocita. Broj blasta u koštanoj srži je normalan ili blago povećan. Serumske razine vitamina B12, proteina koji veže B12 i LDH su povišene proporcionalno broju leukocita. Zbog velikog broja stanica u cirkulaciji (krvi) može doći do lažne hiperkalijemije i hipoglikemije.
Prirodni tijek bolesti
Kronična faza traje 2–4 godine. Za ubrzanu fazu je karakteristična anemija nesrazmjerna aktivnosti bolesti ili liječenju. Broj trombocita pada. Pojavljuju se dodatne citogenetičke abnormalnosti. Raste broj blasta. Obično se unutar 6–8 mjeseci razvije blastna kriza u kojoj prestaje sazrijevanje pa prevladavaju blasti. To je klinička slika akutne leukemije. Oko 50% slučajeva postaje AML, trećina ima morfološke značajke akutne limfocitne leukemije, 10% eritroleukemije, a ostatak su nediferencirane. Preživljenje u blastnoj krizi često je <4 mjeseca.
LIJEČENJE KRONIČNA MIJELOIČNA LEUKEMIJA
Kriterije za terapijski odgovor prikazuje Tbl 65-2. Alogenična transplantacija koštane srži može izliječiti bolest u kroničnoj fazi. Ipak, terapija prve linije je imatinib, molekula koja inhibira djelovanje produkta kimeričnog gena, tj. djelovanje tirozin kinaze. Dnevna peroralna doza od 400 mg dovodi do potpune hematološke remisije u >90% i citogenetske remisije u 76% slučajeva. Ako postoji odgovarajući davatelj, transplantaciju je najbolje izvesti za trajanja potpune remisije. Utvrđeno je nekoliko mehanizama rezistencije na imatinib pa nije vjerojatno da će doći do trajne remisije kada se primjenjuje kao monoterapija. Međutim, kontrolni pregledi nisu dostatni za donošenje čvrstih zaključaka.
TABLICA 65-2 KRITERIJI ZA PROCJENU TERAPIJSKOG ODGOVORA U KRONIČNOJ MIJELOIČNOJ LEUKEMIJI
|
|
Hematološki
|
|
|
Potpun odgovora
|
Leukociti <10.000/μL, normalne morfologije
|
| |
Normalne vrijednosti hemoglobina i trombocita
|
|
Nepotpun odgovor
|
Leukociti ≥10.000/μL
|
|
Citogenetski
|
Postotak metafaza u koštanoj srži s t(9;22)
|
|
Potpun odgovor
|
0
|
|
Djelomičan odgovor
|
≤35
|
|
Manji odgovor
|
36–85b
|
|
Bez odgovora
|
85–100
|
|
Molekularni
|
Prisutnost BCR/ABL transkripta kod RT-PCR
|
|
Potpun odgovor
|
Nijedan (<0.1%)
|
|
Nepotpun odgovor
|
Bilo koji
|
|
aPotpun hematološki odgovor zahtijeva nestanak splenomegalije.
bDo 15% normalnih metafaza ponekad se vidi u vrijeme postavljanja dijagnoze (kada se analizira 30 metafaza).
Kratice: RT-PCR = lančana reakcija polimeraze reverzne transkriptaze.
|
Bolesnici koji više ne reagiraju na imatinib mogu imati povoljan odgovor na druge inhibitore tirozin kinaze kao što su dasatinib (100 mg PO 1× dnevno) ili nilotinib (400 mg PO 2× dnevno). Mutacija T315l u BCR/ABL genu prenosi rezistenciju na sva 3 inhibitora tirozin kinaze. Ponatinib (45 mg/d) je djelotvoran u bolesnika s mutacijom T315l, ali valja biti na oprezu zbog toksičnih učinaka na krvne žile. Alopurinol (300 mg/dan) sprječava uratnu nefropatiju. Jedina kurativna terapija je transplantacija HLA-podudarne alogenične koštane srži. Optimalno vrijeme za transplantaciju nije utvrđeno, ali je transplantacija u kroničnoj fazi učinkovitija nego transplantacija u ubrzanoj fazi ili blastnoj krizi. Transplantacija je najučinkovitija u bolesnika kod kojih se izvrši unutar godine dana od postavljanja dijagnoze. Dugotrajno preživljenje bez bolesti može se postići u 50–60% transplantiranih bolesnika. Infuzija donorskih limfocita može ponovo dovesti do remisije u bolesnika s recidivom. Bolesnicima bez odgovarajućeg davatelja može pomoći transplantacija autolognih matičnih stanica iz periferne krvi. Bolesnike u blastnoj krizi može se liječiti imatinibom, ali terapijski odgovor nije postojan.
MIJELODISPLASTIČNI SINDROMI (MDS)
To su klonski poremećaji stanica koštane srži koji se manifestiraju različitim stupnjem citopenije koje zahvaćaju jednu ili više staničnih loza. Klasifikaciju mijelodisplastičnih sindroma Svjetske zdravstvene organizacije (SZO) prikazuje Tbl. 65-3. Drugi izrazi koji su se upotrebljavali za opisivanje jednog ili više entiteta su preleukemija i oligoblastična leukemija
TABLICA 65-3 KLASIFIKACIJA MIJELODISPLASTIČNIH SINDROMA/NEOPLAZMI SVJETSKE ZDRAVSTVENE ORGANIZACIJE (SZO)
|
|
Naziv
|
SZO: procijenjen postotak pacijenata s MDS-om
|
Periferna krv: ključne karakteristike
|
Koštana srž: ključne karakteristike
|
|
Refraktorne citopenije s displazijom jedne loze (RCUD):
|
|
Refraktorna anemija (RA)
|
10–20%
|
Anemija <1% blasta
|
Eritroidna displazija jedne loze (u ≥10% stanica) <5% blasta
|
|
Refraktorna neutropenija (RN)
|
<1%
|
Neutropenija <1% blasta
|
Granulocitna displazija jedne loze <5% blasta
|
|
Refraktorna trombocitopenija (RT)
|
<1%
|
Trombocitopenija <1% blasta
|
Megakariocitna displazija jedne loze <5% blasta
|
|
Refraktorna anemija s prstenastim sideroblastima (RARS)
|
3–11%
|
Anemija
|
Eritroidna displazija jedne loze ≥15% eritroidnih prekursora su prstenasti sideroblasti <5% blasta
|
| |
|
Bez blasta
|
|
|
Refraktorne citopenije s displazijom više loza (RCMD)
|
30%
|
Citopenija(e) <1% blasta
|
Displazija više loza ± prstenasti sideroblasti <5% blasta
|
| |
|
Bez Auerovih štapića
|
Bez Auerovih štapića
|
|
Refraktorna anemija s viškom blasta, tip 1 (RAEB-1)
|
40%
|
Citopenija(e) <5% blasta
|
Displazija jedne ili više loza
|
| |
|
Bez Auerovih štapića
|
|
|
Refraktorna anemija s viškom blasta, tip 2 (RAEB-2)
|
|
Citopenija(e) 5–19% blasta
|
Displazija jedne ili više loza 10–19% blasta
|
| |
|
± Auerovi štapići
|
± Auerovi štapići
|
|
MDS povezan s izoliranom del(5q)
|
Rijetko
|
Anemija
|
Izolirana delecija kromosoma 5q31
|
| |
|
Trombociti normalni ili povišeni
|
Anemija; hipolobulirani megakariociti
|
| |
|
<1% blasta
|
<5% blasta
|
|
MDS dječje dobi, uključujući refraktornu citopeniju dječje dobi (privremeno) (RCC)
|
1 %
|
Pancitopenija
|
<5% blasta u koštanoj srži za RCC
|
| |
|
|
Koštana srž obično hipocelularna
|
|
MDS, neklasificiran (MDS-U)
|
?
|
Citopenija
≤1% blasta
|
Ne uklapa se u ostale kategorije
Displazija
<5% blasta
Ako nema displazije, kariotip povezan s MDS-om
|
|
Napomena: Ako je broj blasta u perifernoj krvi 2–4%, dijagnoza je RAEB-1 čak i ako je broj blasta u koštanoj srži <5%. Ako su prisutni Auerovi štapići, SZO uzima u obzir dijagnozu RAEB-2 ako je udio blasta <20% (čak i <10%), AML ako je barem 20% blasta. Za sve podtipove, broj monocita u perifernoj krvi je <1 × 109/L. Bicitopenija se može naći u RCUD podtipovima, ali pancitopenija s displazijom jedne loze u koštanoj srži trebala bi se klasificirati kao MDS-U. MDS povezan s terapijom (t-MDS), bilo zbog alkilirajućih agenasa, topoizomeraze II (t-MDS/t-AML) u klasifikaciji AML-a i prekursorskih bolesti SZO-a. Popis u ovoj tablici isključuje kategorije preklapanja MDS/mijeloproliferativne neoplazme, kao što su kronična mijelomonocitna leukemija, juvenilna mijelomonocitna leukemija i privremeni entitet RARS s trombocitozom.
Kratice: MDS= mijelodisplastični sindrom.
|
Incidencija i etiologija
Godišnje se javlja oko 3000 novih slučajeva, uglavnom u ljudi >50 godina (srednja dob, 68). Kao i kod AML-a, izloženost benzenu, radijaciji i citostaticima može dovesti do MDS-a. Poremećaji kromosoma javljaju se u do 80% slučajeva, uključujući deleciju dijela ili cijelog kromosoma 5, 7 i 9 (rjeđe 20 ili 21) i dodavanje (adiciju) dijela ili cijelog kromosoma 8. Mutacije u genima povezanim s uklanjanjem introna (splicing) transkripcijske RNK, kao što je SF3B1 imaju povoljniju prognozu. Mutacije u genima koji su često uključeni u AML, kao što su RUNX i ASXL1 imaju lošiju prognozu.
Kliničke i laboratorijske značajke
Simptomi ovise o zahvaćenim staničnim lozama. Oko 85% bolesnika je anemično, 50% ima neutropeniju, a oko trećine ima trombocitopeniju. Patološke značajke MDS-a su celularna koštana srž s različitim stupnjem citološke atipije uključujući usporeno sazrijevanje jezgre, abnormalno sazrijevanje citoplazme, nakupljanje prstenastih sideroblasta (mitohondriji puni željeza koji okružuju staničnu jezgru), megakariocite s jednim ili dva režnja, mikromegakariocite i povećan broj mijeloblasta. U Tbl 65-3 navedene su karakteristične značajke za identifikaciju pojedinih entiteta. Prognozu određuje % blasta u koštanoj srži, kariotip i zahvaćene stanične loze. Međunarodni prognostički bodovni sustav prikazuje Tbl 65-4.
TABLICA 65-4 MEĐUNARODNI PROGNOSTIČKI SUSTAV BODOVANJA (IPSS)
|
|
Prognostička varijabla
|
Bodovi
|
|
0
|
0.5
|
1
|
1.5
|
2
|
|
Blasti u koštanoj srži (%)
|
<5%
|
5–10%
|
|
11–20%
|
21–30%
|
|
Kariotipa
|
Dobar
|
Srednji
|
Loš
|
|
|
|
Citopenijab (zahvaćene loze)
|
0 ili 1
|
2 ili 3
|
|
|
|
|
Rizik
|
Bodovi
|
|
|
|
|
|
Niski
|
0
|
|
|
|
|
|
Srednji-1
|
0.5–1
|
|
|
|
|
|
Srednji-2
|
1.5–2
|
|
|
|
|
|
Visoki
|
≥2.5
|
|
|
|
|
|
aDobar, normalan, -Y, del(5q), del(20q); loš, kompleksan (≥3 abnormalnosti) ili poremećaji kromosoma 7; srednji, svi ostali poremećaji.
bCitopenije definirane kao Hb <100 g/L, broj trombocita <100.000/μL, apsolutni broj neutrofila <1500/μL.
|
LIJEČENJE MIJELODISPLASTIČNI SINDROMI
Alogenična transplantacija koštane srži jedina je kurativna terapija i može izliječiti 60% transplantiranih bolesnika. Međutim, većina je bolesnika s MDS-om prestara za transplantaciju. 5-Azacitidin (75 mg/m2 dnevno × 7, svaka 4 tjedna) može usporiti transformaciju u AML za 8–10 mjeseci. Decitabin (15 mg/m2 u kontinuiranoj infuziji, 3× dnevno, ×3 kure, svakih 6 tjedana) može inducirati terapijski odgovor koji prosječno traje 1 godinu dana u 20% slučajeva. Lenalidomid (10 mg/d), analog talidomida s manje učinaka na CNS, dovodi do toga da značajan dio bolesnika sa sindromom 5q– postane neovisan o transfuziji. Bolesnici s niskom razinom eritropoetina mogu imati terapijski odgovor na eritropoetin, a manji broj bolesnika s neutropenijom odgovor na faktor stimulacije rasta granulocita. Potporna terapija je temelj liječenja.
MIJELOPROLIFERATIVNI SINDROMI
Tri glavna mijeloproliferativna sinroma su policitemija vera, idiopatska mijelofibroza i esencijalna trombocitoza. Svi su klonalni poremećaji hematopoetskih matičnih stanica i svi su povezani s mutacijom JAK2 kinaze (V617F) koja dovodi do aktivacije kinaze. Mutacija je uočena u 90% bolesnika s policitemijom verom i ~45% bolesnika s idiopatskom mijelofibrozom i esencijalnom trombocitozom.
POLICITEMIJA VERA
Najčešći mijeloproliferativni sindrom, a odlikuje se povećanom masom eritrocita, masivnom splenomegalijom i kliničkim manifestacijama vezanim uz povećanu viskoznost krvi, uključujući neurološke simptome (vertigo, tinitus, glavobolju, smetnje vida) i trombozu (infarkt miokarda, moždani udar, bolest perifernih krvnih žila; rjeđe mezenterijalnih i jetrenih). Mora se razlučiti od drugih uzroka povećanja mase eritrocita (Pogl. 45). To se najlakše postiže određivanjem razine eritropoetina u serumu. Policitemija vera je povezana s vrlo niskom razinom eritropoetina. Kada se radi o drugim uzrocima eritrocitoze razina eritropoetina je povišena. Testovi za mutaciju JAK2(V617F) danas su u svakodnevnoj upotrebi. Bolesnici se učinkovito liječe venepunkcijama. Nekim je bolesnicima potrebno učiniti splenektomiju za kontrolu simptoma, a onima s jakim svrbežem mogu koristiti psoralen i UV svijetlo. U 20% bolesnika razvije se mijelofibroza, a akutna leukemija u <5%. Inhibitor JAK1 i JAK2, ruksolitinib je u fazi testiranja.
IDIOPATSKA MIJELOFIBROZA
Za ovaj rijedak entitet karakteristična je fibroza koštane srži, mijeloidna metaplazija s ekstramedularnom hematopoezom i splenomegalija. Pregled krvnog razmaza otkriva eritrocite u obliku suze, eritrocite s jezgrom i ponešto ranih oblika granulocita, uključujući promijelocite. Međutim, mnogi entiteti mogu dovesti do fibroze koštane srži i ekstramedularne hematopoeze pa se dijagnoza primarne idiopatske mijelofibroze postavlja tek kada se isključe brojni potencijalni uzroci. Diferencijalno dijagnostički u obzir dolaze slijedeće bolesti: KML, policitemija vera, Hodgkinova bolest, metastaze karcinoma u koštanu srž (osobito dojke i prostate), infekcije (naročito granulomatozne), te leukemija vlasastih stanica. U pravilu se primjenjuje potporna terapija. Novi inhibitori JAK2 i telomeraze pokazali su djelotvornost u smanjenju splenomegalije i fibroze koštane srži u nekim slučajevima. Međutim, nijedna studija još nije pokazala da određena farmakoterapija poboljšava (produljuje) preživljenje. Bolesnici koji nemaju JAK2 mutacije često imaju CALR mutacije.
ESENCIJALNA TROMBOCITOZA
Obično se otkriva slučajno prilikom rutinskog određivanja broja trombocita u sklopu laboratorijskih pretraga u asimptomatskih bolesnika. Poput mijelofibroze, mnoga stanja mogu dovesti do povećanja broja trombocita (trombocitoze). Zato se dijagnoza postavlja metodom isključivanja. Broj trombocita mora biti >500.000/µL, a poznate uzroke trombocitoze treba isključiti uključujući KML, manjak željeza, splenektomiju, malignitet, infekciju, krvarenje, policitemiju veru, mijelodisplaziju i oporavak nakon deficijencije vitamina B12. Premda je obično asimptomatska, bolesnike treba liječiti ako razviju migrensku glavobolju, tranzitornu ishemičnu ataku (TIA), ili neku drugu manifestaciju krvarenja ili tromboze. Interferon-α je učinkovita terapija, jednako kao i anagrelid i hidroksiureja. Izoliranu trombocitozu (visoki apsolutni broj trombocita) bez drugih simptoma ne treba liječiti. U 80% slučajeva nalazimo JAK2 i CALR mutacije; u oko 10% slučajeva MPL mutacije.
Opširnije vidi u Young NS: Aplastic Anemia, Myelodysplasia, and Related Bone Marrow Failure Syndromes, Pogl. 130, str. 662; Spivak JL: Polycythemia Vera and Other Myeloproliferative Diseases, Pogl. 131, str. 672; i Marcucci G, Bloomfield CD: Acute Myeloid Leukemia, Pogl. 132, str. 678; Kantarjian H, Cortes J: Chronic Myeloid Leukemia, Pogl. 133, str. 687, u HPIM-19.