Urednik sekcije: prof. dr. sc. Srđan Novak, dr. med.
Prijevod: prof. dr. sc. Srđan Novak, dr. med.
DEFINICIJA
Amiloidoza je pojam za skupinu proteinopatija (poremećaji prostorne strukture proteina, tj. nenormalna trodimenzionalna struktura proteina) što dovodi do taloženja netopljivih polimernih amiloidnih vlakana, većinom u izvanstaničnim prostorima tkiva i organa. Klinička slika ovisi o anatomskoj raspodjeli i količini odloženog amiloida, a kreće se od lokalnih depozita malog značaja do zahvaćanja praktički svih organskih sustava s ozbiljnim patofiziološkim promjenama.
KLASIFIKACIJA
Amiloidoze se razlikuju s obzirom na biokemijske odlike bjelančevina koje tvore amiloidna vlakna i klasificiraju se prema tome jesu li sistemske ili lokalizirane, stečene ili naslijeđene, te prema kliničkoj slici. Prihvaćena nomenklatura je AX gdje A označava amiloidozu a X je bjelančevina u vlaknu (Tbl. 137-1, str. 720, HPIM-19).
- AL (imunoglobulini lakih lanaca): Primarna amiloidoza; najčešći oblik sistemske amiloidoze; nastaje zbog poremećaja klona B stanica, obično u sklopu multiplog mijeloma.
- AA (serumski amiloid A): sekundarna amiloidoza; može se javiti uz gotovo svako kronično upalno stanje [npr. RA, SLE, sindromi periodične groznice kao što je obiteljska Mediteranska groznica (OMG), Crohnova bolest] ili uz kronične infekcije.
- AF (obiteljske amiloidoze): veliki broj različitih oblika koji se dominantno nasljeđuju, a vezani su za mutacije koje potiču pogrešno nabiranje bjelančevina i oblikovanje vlakana; najčešće zbog transtiretina.
- Aβ2M: građen od β2-mikroglobulina; nastaje u terminalnom stadiju dugotrajne bubrežne bolesti.
- Lokalizirana ili amiloidoza jednog organa: najčešći oblik je Aβ nađen u oboljelih od Alzheimerove bolesti a nastaje zbog abnormalne proteolitičke obrade amiloidnog prekursorskog proteina (APP).
KLINIČKA SLIKA
Kliničke manifestacije su raznolike i ovise o biokemijskoj strukturi bjelančevina amiloidnih vlakana. Bolest često zahvaća:
- Bubreg: kod AA i AL amiloidoze; proteinurija, nefroza, azotemija.
- Jetru: kod AA, AL i AF; hepatomegalija.
- Kožu: karakteristično za AL amiloidozu, ali se može vidjeti i kod AA amiloidoze; voštane papule.
- Srce: karakteristično za AL i obiteljsku amiloidozu (AF); kongestivno zatajenje srca, kardiomegalija, aritmije.
- GI trakt: karakteristična lokalizacija za sve oblike amiloidoze; GI opstrukcija ili ulceracije, krvarenje, gubitak proteina, proljev, makroglosija, poremećaj motiliteta jednjaka.
- Zglobove: većinom kod AL; često u sklopu mijeloma; periartikularni depoziti amiloida, “znak ramenog jastučića”: tvrdi depoziti amiloida u mekom tkivu oko ramenog zgloba, simetrični artritis ramena, ručnih zglobova, zglobova koljena i šaka.
- Živčani sustav: većinom je zahvaćen kod nasljednih obiteljskih amiloidoza (AF); periferna neuropatija, posturalna hipotenzija, demencija. Sindrom karpalnog kanala se može javiti kod AL i Aβ2M oblika amiloidoze.
- Dišni sustav: AL amiloidoza može zahvatiti donje dišne putove; lokalizirani oblik amiloidoze može dovesti do opstrukcije duž gornjih dišnih putova.
DIJAGNOZA
Dijagnoza se postavlja identifikacijom amiloidnih depozita u bioptatu zahvaćenih tkiva i tipizacijom amiloida (Sl. 167-1). Bojenje Kongo crvenilom aspirata abdominalnog masnog tkiva će otkriti naslage amiloida kod >80% bolesnika sa sistemskom amiloidozom.
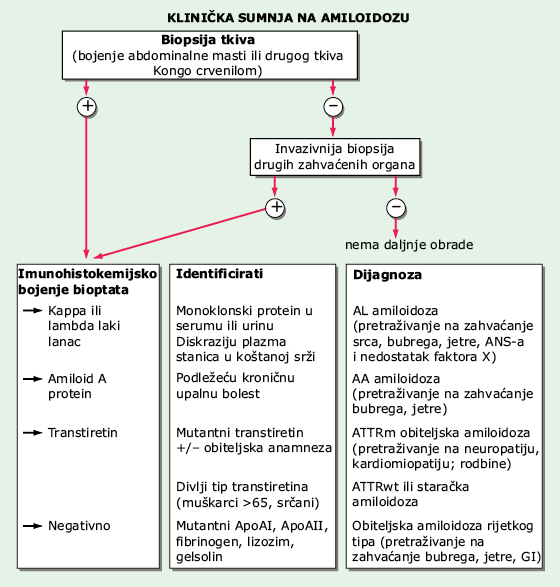
SLIKA 167-1 Algoritam za postavljenje dijagnoze i određivanje tipa amiloidoze: Klinička sumnja: neobjašnjiva nefropatija, kardiomiopatija, neuropatija, enteropatija, artropatija i makroglosija. ApoAI = apolipoprotein AI, ApoAII = apolipoprotein AII, GI = gastrointestinalno.
PROGNOZA
Ishod je promjenljiv i ovisi o tipu i proširenosti amiloidoze. Prosječno preživljenje kod AL amiloidoze bez liječenja iznosi ~1–2 godine; zahvaćanje srca je vodeći uzrok smrti s prosječnim preživljavanjem ~6 mjeseci bez liječenja.
LIJEČENJE AMILOIDOZA
Za AL, trenutne terapije usmjerene su na klonove plazma stanica koštane moždine pristupom koji se primjenjuju kod liječenja multiplog mijeloma. Nakon visoke IV doze melfalana slijedi autologna transplantacija matičnih stanica koja dovodi do kompletnog hematološkog odgovora u ~40% slučajeva, ali samo 50% oboljelih ima pravo na takav agresivni tretman, a peritransplantacijska smrtnost veća je nego kod ostalih hematoloških bolesti zbog oštećene funkcije organa. Liječenje AA je usmjereno na kontrolu osnovnog upalnog stanja. Kolhicin (1,2–1,8 mg/d) je standardna terapija za OMG, ali nije se pokazala korisnom za AA drugih uzroka. Inhibitori TNF i antagonisti interleukina-1 mogu biti učinkoviti kod sindroma povezanih s porastom citokina. Ispituje se eprodizat koji je dizajniran da prekida stvaranje fibrila. Za neke je oblike nasljedne obiteljske amiloidoze (AF) važno genetsko savjetovanje, a transplantacija jetre je uspješan oblik liječenja.
Opširnije vidi u Seldin DC, Berk JL: Amyloidosis, Pogl. 137, str. 719, u HPIM-19.