ŠEĆERNA BOLEST I DRUGI METABOLIČKI POREMEĆAJI
Šećerna bolest
Akutne komplikacije šećerne bolesti
Kronične komplikacije šećerne bolesti
Posebna stanja vezana uz šećernu bolest
Hipoglikemija
Poremećaji metabolizma lipida (hiperlipidemije)
Poremećaji metabolizma mokraćne kiseline
Poremećaji metabolizma porfirina (porfirije)
Poremećaji metabolizma aminokiselina
Poremećaji metabolizma željeza
Poremećaji metabolizma bakra
Poremećaji koštanoga metabolizma
Lizosomske bolesti nakupljanja
Bolesti nakupljanja glikogena (glikogenoze)
Poremećaji prehrane: pretilost, metabolički sindrom i malnutricija
Literatura
Šećerna bolest predstavlja jednu od najraširenijih kroničnih bolesti u odrasloj populaciji. Prema podacima Međunarodne dijabetičke federacije (IDF) trenutno u svijetu 463 milijuna ljudi boluje od šećerne bolesti. U nastavku teksta opisane su osnove regulacije glikemije, definicija, klasifikacija, etiopatogenetski mehanizmi, klinička slika bolesti te dijagnostički i terapijski pristup oboljelima od šećerne bolesti.
REGULACIJA GLIKEMIJE I FIZIOLOGIJA INZULINA
Glikemija. Koncentracija glukoze u plazmi strogo je regulirana i održavana u uskom rasponu. Ravnoteža se održava između ulaska glukoze u cirkulaciju iz jetre ili probavnoga sustava nakon obroka te utilizacije glukoze u perifernim tkivima. Stalna koncentracija glukoze posebno je bitna za mozak, s obzirom na to da je glukoza esencijalni energent neuronima koji u energetske svrhe ne mogu oksidirati masne kiseline, nego se oslanjaju na glukozu kao glavni metabolički energent. Kada kod zdravih osoba između uzimanja obroka dođe do smanjene apsorpcije glukoze iz probavnoga sustava, dolazi do povećanoga otpuštanja glukoze iz jetre kao odgovora na smanjenje koncentracije inzulina i povećanja koncentracije kontraregulatornih hormona, posebno glukagona. Glukoza u jetri nastaje razgradnjom glikogena (glikogenoliza) ili stvaranjem glukoze iz neugljikohidratnih supstrata kao što su glicerol i aminokiseline, procesom koji se zove glukoneogeneza. Nakon obroka, kada dolazi do pojačane apsorpcije glukoze iz probavnoga sustava, dolazi i do povećanoga lučenja inzulina koji ulazi u portalnu cirkulaciju, a zatim i jetru i ostala periferna tkiva. Inzulin smanjuje koncentraciju glukoze stimuliranjem prijenosa glukoze u stanice te supresijom produkcije glukoze u jetri. Inzulin stimulira i lipogenezu te inhibira lipolizu.
Sinteza inzulina. Gen koji kodira humani inzulin nalazi se na kratkom kraku jedanaestoga kromosoma. Inzulin se inicijalno sintetizira u β-stanicama Langerhansovih otočića kao jednolančani polipeptid sačinjen od 86 aminokiselina. Potom dolazi do odcjepljivanja C-peptida te do konačne formulacije molekule inzulina kao dvolančanoga polipetida kojeg čini 51 aminokiselina. I inzulin i C-peptid potom se skladište u granule iz kojih će se izlučiti u portalnu cirkulaciju, a to izlučivanje inzulina i C-peptida je ekvimolarno. Oni iz portalne cirkulacije dospijevaju u jetru gdje se velik dio inzulina veže za odgovarajuće receptore i biva metaboliziran već pri prvome prolasku kroz jetru. C-peptid u velikoj mjeri izbjegava promjene u jetri, pa vrijednosti C-peptida u perifernoj krvi zapravo bolje odražavaju endogenu sekreciju inzulina od same koncentracije inzulina.
Izlučivanje inzulina. Serumska koncentracija glukoze glavni je regulator lučenja inzulina. Sam proces lučenja inzulina započinje ulaskom glukoze u β-stanice Langerhansovih otočića putem GLUT2 transportnoga proteina. Nakon toga ulaska dolazi do fosforilacije glukoze putem enzima glukokinaze te ona biva metabolizirana. Kao završni produkt toga metabolizma javlja se nakupljanje ATP-a koji inhibira aktivnost o ATP-u ovisnih kalijskih kanala u membrani β-stanica što dovodi do depolarizacije stanice. Uslijed depolarizacije membrane β-stanica, dolazi do otvaranja kalcijskih kanala i utoka kalcija u citoplazmu, a to potiče degranulaciju i oslobađanje inzulina i C-peptida. Nakon odgovarajućega poticaja ponajprije se luči već stvoren inzulin pohranjen u granulama u citoplazmi, no ako poticaj traje dulje, dolazi do pojačanoga stvaranja inzulina de novo i nastavka lučenja. Samo lučenje inzulina pulsatilnoga je karaktera. Jačina inzulinske sekrecije određena je koncentracijom glukoze u serumu, tako da vrijednosti glukoze iznad 3,9 mmol/L potiču sintezu inzulina. Također, jačina inzulinske sekrecije ovisi i o načinu unosa glukoze u organizam: oralnim unosom glukoze postiže se jači odgovor β-stanica i jače lučenje inzulina nego intravenskim unosom glukoze, što se tumači izostankom dodatne stimulacije lučenja inzulina putem inkretina, proteina kojeg luči probavni sustav kao odgovor na unos glukoze (GLP-1, GIP...). Inkretine stvaraju i otpuštaju neuroendokrine stanice probavnoga sustava nakon unosa hrane te imaju takav učinak da amplificiraju glukozom stimulirano lučenje inzulina te suprimiraju sekreciju glukagona. Glukagonu sličan peptid (GLP-1, engl. glucagon-like peptide 1) najpotentniji je inkretin kojeg luče L-stanice tankoga crijeva, a potiče lučenje inzulina samo kada je koncentracija glukoze u serumu povišena.
Učinak inzulina. Jednom izlučen inzulin dospijeva u sustav vene porte odakle oko 50 % inzulina biva uklonjeno i degradirano od strane jetre već kod prvoga prolaska. Ostatak inzulina dospijeva u sistemsku cirkulaciju gdje se veže na inzulinske receptore koji se nalaze na ciljnim stanicama. Primarna ciljna tkiva inzulina su jetra, stanice skeletne muskulature te masne stanice. Inzulinski receptor je heterodimer koji se sastoji od dva α i dva β-lanca povezana disulfidnim vezama. Α-lanci nalaze se na vanjskoj strani membrane i predstavljaju mjesto na koje se veže inzulin, dok su β-lanci smješteni iznutra i povezani su s tirozin kinazom koja je ključna za funkciju receptora. Vezanjem inzulina za inzulinski receptor dolazi do stimulacije aktivnosti unutarstanične tirozin kinaze koja dovodi do autofosforilacije receptora i do aktivacije unutarstaničnih signalnih molekula pod nazivom IRS (engl. insulin receptor substrates). One pokreću različite reakcije koje dovode do konačnih učinaka inzulina na stanice. Jedna je od tih reakcija i aktivacija fosfatidilinozitol-3-kinaze koja stimulira translokaciju trasportera glukoze GLUT4 na površinu stanice. Preko njega glukoza ulazi u stanice skeletnih mišića te u masne stanice. Aktivacija drugih signalnih puteva dovodi do poticanja sinteze glikogena, sinteze proteina te lipogeneze.
Metabolički učinci inzulina. Metabolički učinci inzulina su brojni, s obzirom na to da on, osim na metabolizam ugljikohidrata, utječe i na metabolizam lipida i proteina. Učinci mogu biti anabolički i katabolički. Na metabolizam ugljikohidrata utječe tako da potiče transport glukoze u stanice jetre, skeletnih mišića i masne stanice, stimulira fosforilaciju glukoze intracelularno, potiče glikogenezu, stimulira aktivnost piruvat dehidrogenaze te koči glukoneogenezu i glikogenolizu. Na metabolizam lipida utječe tako da potiče sintezu triglicerida te masnih kiselina, potiče aktivnost lipoprotein lipaze u masnom tkivu, koči lipolizu i ketogenezu te inhibira aktivnost lipoprotein lipaze u mišićima i oksidaciju masnih kiselina u jetri. Na metabolizam proteina utječe tako da potiče transport aminokiselina i sintezu proteina, a koči degradaciju (razgradnju) proteina.
Uloga kontraregulatornih hormona. Kontraregulatorni hormoni su hormoni čiji je učinak suprotan učinku inzulina. To su glukagon, hormon rasta, kortizol i katekolamini. Najznačajniji učinak ima glukagon. On se normalno stvara i luči iz α stanica Langerhansovih otočića kao odgovor na hipoglikemiju i aktivaciju autonomnoga živčanog sustava. Glavni učinak ostvaruje u jetri gdje stimulira glikogenolizu, glukoneogenezu i ketogenezu putem mehanizama ovisnih o adenozin-monofosfatu. Normalno, lučenje glikogena je inhibirano hiperglikemijom i inzulinom. Ipak, u oba tip šećerne bolesti (tipa 1 i tip 2), unatoč hiperglikemiji i povišenoj serumskoj koncentraciji inzulina, nalazimo i povišene koncentracije glukagona. Hormon rasta kojega luči prednji režanj hipofize također je povišen kod bolesnika sa šećernom bolesti. Glavni učinak hormona rasta ostvaruje se u perifernim tkivima gdje dovodi do lipolize i smanjenoga iskorištavanja glukoze. Kortizol i katekolamini su takozvani hormona stresa koji se luče u raznim bolestima i drugim stresnim stanjima što može dodatno pogoršati hiperglikemiju bolesnika sa šećernom bolešću.
DEFINICIJA I KLASIFIKACIJA ŠEĆERNE BOLESTI
Šećerna bolest (lat. diabetes mellitus) kronični je poremećaj metabolizma ugljikohidrata, proteina i masti koji nastaje uslijed apsolutnoga ili relativnog manjka inzulina ili uslijed inzulinske rezistencije. Šećerna bolest posljedica je kompleksnih interakcija između genetskih i okolišnih čimbenika. Bez obzira na uzrok, glavno obilježje šećerne bolesti je hiperglikemija, a neliječena i loše regulirana šećerna bolest povezana je s oštećenjem brojnih organa što danas predstavlja sve veći klinički i javnozdravstveni problem.
Šećernu bolest klasično dijelimo u četiri skupine: šećerna bolest tip 1 i tip 2, gestacijska šećerna bolest te ostali, specifični oblici šećerne bolesti. Šećerna bolest tip 1 dijeli se u dva podtipa: autoimuni i idiopatski. Autoimuna šećerna bolest tip 1 primarno je uzrokovana autoimunom destrukcijom β-stanica i karakterizirana je apsolutnim manjkom inzulina, a javlja se u mlađoj životnoj dobi (tzv. imunoposredovana šećerna bolest tip 1), dok je idiopatska šećerna bolest tip 1 daleko rjeđi oblik i čini oko 5 % šećerne bolesti tip 1. U ovom obliku ne nalazimo parametre koji upućuju na autoimunu destrukciju β-stanica i ne zna se točan uzrok, zbog čega se i naziva idiopatska šećerna bolest tip 1. Šećerna bolest tip 2 nastaje u starijoj životnoj dobi, a ponajprije je uzrokovana inzulinskom rezistencijom (neosjetljivost perifernih tkiva na inzulin) te sekundarnim razvojem relativnoga manjka inzulina. Gestacijska šećerna bolest zasebni je klinički entitet karakteriziran pojavom hiperglikemije uzrokovane različitim fiziološkim promjenama u trudnoći. Ostali, specifični oblici šećerne bolesti su heterogena skupina bolesti i stanja koja su jasno povezana s nastankom šećerne bolesti. Tu klasifikaciju dala je ADA (engl. America diabetes association) i široko je prihvaćena u svijetu. Bez obzira na to koliko klasifikacija bila detaljna, uvijek postoji „siva zona“ u kojoj je otežano razlučivanje tip i uzroka šećerne bolesti.
EPIDEMIOLOGIJA ŠEĆERNE BOLESTI
Šećerna bolest jedan je od najvećih javnozdravstvenih problema, kako u svijetu, tako i u nas. Međunarodna dijabetička federacija (IDF, engl. International diabetes federation) procjenjuje da u svijetu od te bolesti boluje 463 milijuna osoba (svaka jedanaesta odrasla osoba) s tenedencijom stalnoga porasta oboljelih. Smatra se također da gotovo polovica osoba koje boluju od šećerne bolesti još nema postavljenu dijagnozu. Na liječenje šećerne bolesti i njezinih komplikacija otpada 12 % svjetskih troškova u zdravstvu.
Prema Nacionalnome registru osoba sa šećernom bolešću u Republici Hrvatskoj 2019. godine bilo je više od 300 000 odraslih osoba s dijagnosticiranom šećernom bolesti. Međutim, procjenjuje se da je ukupan broj osoba sa šećernom bolesti u Republici Hrvatskoj veći od 450000. Najučestaliji tip šećerne bolesti je tip 2 i čini preko 90 % slučajeva. Šećerna bolest nalazi se među deset vodećih uzroka smrti u Republici Hrvatskoj.
ETIOPATOGENEZA ŠEĆERNE BOLESTI tip 1
Šećerna bolest tip 1 nastaje kao rezultat interakcije genetskih, okolišnih te imunoloških čimbenika. Radi se o autoimunom oštećenju β-stanica kod genetski predisponiranih pojedinaca, vjerovatno potaknutom nekim vanjskim, danas još nepoznatim čimbenikom. Bolest se razvija mjesecima i godinama uz postupno razaranje β-stanica. Taj period karakterizira inzulinitis, odnosno infiltracija otočića upalnim infiltratom kojega ponajprije čine aktivirani makrofagi, T-limfociti, NK-stanice te B-limfociti. Hiperglikemija i manifestna bolest nastupaju kada je razoreno više od 70 - 80 % β-stanica, kada se više ne može izlučiti dovoljno inzulina da bi se održala normalna glikemija. Te se promjene obično zbivaju u djetinjstvu ili pubertetu. Prijelaz iz stabilne u nestabilnu fazu bolesti često je izazvan pojačanim metaboličkim zahtjevima kao što su infekcije ili pubertet. Šećerna bolest tip 1 često je udružena i s drugim autoimunim bolestima kao što su autoimune bolesti štitnjače, Addisonova bolest, perniciozna anemija ili vitiligo.
Genetska predispozicija. Danas je jasno da postoji genetska predispozicija za razvoj šećerne bolesti tip 1. Ipak, samo postojanje određenih genetskih značajki ne znači ujedno i sigurno obolijevanje, nego u tome važnu ulogu imaju i određeni drugi čimbenici. To je poznato zbog činjenice da je istodobna pojavnost šećerne bolesti tip 1 kod identičnih blizanaca između 40 i 60 %, što ukazuje na druge čimbenike u samoj patogenezi bolesti. Smatra se da više od dvadeset različitih regija humanoga genoma pokazuje određenu povezanost s razvojem šećerne bolesti tip 1, a HLA regija, smještena na kratkom kraku šestoga kromosoma, najpovezivija je s razvojem ove bolesti. Većina osoba sa šećernom bolesti tip 1 ima HLA DR3 i/ili HLA DR4 haplotip. Preciznija istraživanja pokazuju da su haplotipovi DQA1*0301, DQB1*0302 i DQB1*0201 snažno povezani s razvojem bolesti. Ti haplotipovi prisutni su kod više od 40 % djece sa šećernom bolesti tip 1, za razliku od zdrave djece kod koje su prisutni u samo 2 %. Poznati su i drugi geni koji se povezuju s razvojem bolesti, kao što su CD25, PTPN22, IL2RA i drugi. To su geni koji sudjeluju u procesima razvoja T-limfocita te u regulaciji imunoloških procesa. Ti su geni, osim što su povezani sa šećernom bolesti tip 1, povezani i s razvojem drugih imunološki posredovanih bolesti kao što su celijakija, bolesti štitnjače i dr. Opisani su i haplotipovi čija je prisutnost povezana s manjom učestalošću ove bolesti, zbog čega se smatraju protektivnima. Takvi su haplotipovi DQA1*0102 i DQB2*0602, i oni su ekstremno rijetki kod bolesnika sa šećernom bolesti tip 1 (manje od 1 %).
Okolišni čimbenici. Do danas nije poznat točan čimbenik (okidač) koji će kod genetski predisponiranoga pojedinca uzrokovati aktivaciju imunološkoga zbivanja koje će dovesti do destrukcije β-stanica. Više je čimbenika koji se mogu uzeti u obzir na temelju rezultata različitih studija. Najčešće se spominju virusne infekcije, prehrana i stres. Virusne infekcije mogu doprinijeti izmjeni imunogenosti β-stanica, što može pripomoći aktivaciji autoimunoga procesa. Najčešće se spominju virus zaušnjaka, Coxsackie virus, retrovirusi, rubeola virus, citomegalovirus i Epstein-Barrov virus. Prehrambene navike također mogu biti važne za razvoj bolesti. Smatra se da različiti nitrozamini koje nalazimo u suhomesnatim proizvodima mogu imati potencijalno toksičan učinak na β-stanice. Isti učinak mogu imati proteini u kravljem mlijeku. Rano uvođenje kravljega mlijeka u prehranu stvara veći rizik za razvoj šećerne bolesti tip 1 kod djece, u odnosu na dojenu djecu.
Imunološki čimbenici. Da se u šećernoj bolesti tip 1 radi o imunološki posredovanom oštećenju β-stanica govore brojne studije i istraživanja. U prilog tome govore sljedeće značajke oboljelih: 1) prisutnost protutijela na β-stanice; 2) prisutnost aktiviranih T-limfocita u Langerhansovim otočićima, peripankreatičkim limfnim čvorovima i sistemskoj cirkulaciji; 3) mogućnost stimulacije proliferacije T-limfocita uz dodatak proteina dobivenih iz β-stanica u in vitro uvjetima te 4) povećana prisutost citokina u Langerhansovim otočićima i sistemskoj cirkulaciji. U većine bolesnika nalazimo antitijela usmjerena protiv β-stanica (ICS, engl. islet cell autoantibodies). Molekule na koje imunološki sustav reagira proizvodnjom autoantitijela su različite: inzulin, izoforme glutaminske dekarboksilaze (GAD65, GAD 67), protein sekretornih granula (ICA512/IA-2) te specifični cinkov transporter (ZnT-8). Komercijalno se određuju jedino GAD65 protutijela. Navedena autoantijela prisutna su kod 85 % bolesnika sa šećernom bolesti tip 1 i 5 do 10 % bolesnika sa šećernom bolesti tip 2. Iako se nalaze kod većine oboljelih, ta se autoantitijela ne smatraju bitnima u samome procesu destrukcije β-stanica. Destrukcija β-stanica posredovana je izravnim citotoksičnim djelovanjem aktiviranih CD8+ T-limfocita te brojnim citokinima koje nalazimo u upalnom infiltratu. Nakon što su β-stanice uništene, dolazi do smirivanja upalnoga procesa, Langerhansovi otočići atrofiraju, a postupno nestaju i navedeni imunološki markeri. Na animalnim su modelima iskušani različiti načini prevencije šećerne bolesti tip 1 putem raznih utjecaja na imunološki sustav (primjena anti-CD3 monoklonalnoga protutijela, anti-B limfocitnih monoklonalnih protutijela, imunosupresija) s različitim uspjehom.
Metabolički poremećaji. Razvoj šećerne bolesti tip 1 je polagan, a metabolički poremećaji nastupa kada je uništeno više od 70 od 80 % β-stanica jer tada produkcija inzulina prestaje biti dovoljna za održavanje normalnih metaboličkih procesa. Hiperglikemija dovodi do dodatnoga toksičnog učinka na β-stanice što pridonosi još bržem uništenju β-stanica i potpunom prestanku stvaranja inzulina. Hiperglikemija dovodi do glikozurije i posljedične dehidracije (smanjenje intravaskularnoga volumena), a to dovodi do sekundarnoga hiperaldosteronizma. Uslijed manjka inzulina dolazi i do pojačane lipolize i proteolize, posljedica čega je gubitak na tjelesnoj masi, pojačane glukoneogeneze i ketogeneze. Kada produkcija ketonskih tijela nadiđe razinu sposobnosti njihova metabolizma, oni se nakupljaju te dovode do razvoja ketoacidoze (ketonska tijela su po kemijskom sastavu kiseline).
ETIOPATOGENEZA ŠEĆERNE BOLESTI tip 2
Etiopatogeneza šećerne bolesti tip 2 nešto je složenija i uključuje dva osnovna obilježja, a to su inzulinska rezistencija i oštećena funkcija β-stanica koja dovodi do relativnoga manjka inzulina. Primarno se razvija inzulinska rezistencija što gušterača pokušava nadokanditi pojačanom produkcijom inzulina, stoga se u prvoj fazi bolesti javlja hiperinzulinemija. S obzirom na to da takvo stanje duže traje, dolazi do zamora β-stanica što vodi prema njihovom iscrpljenju i smanjenoj funkciji, pa dolazi do postupnoga smanjenja lučenja inzulina. Kada izlučivanje inzulina padne ispod razine potrebne da bi se održala euglikemija u stanju inzulinske rezistencije, dolazi do razvoja hiperglikemije. Karakteristično se šećerna bolest razvija u odrasloj životnoj dobi. Prirodni tijek i razvoj šećerne bolesti tip 2 prikazan je na Slici 6.
Genetska predispozicija. Šećerna bolest tip 2 također pokazuje snažnu vezu s genetskim promjenama. Istodobna pojavnost šećerne bolesti tip 2 kod identičnih blizanaca iznosi 70 do 90 %, što ukazuje na jaku genetsku podlogu razvoja bolesti. Osoba čiji roditelj ima šećernu bolest tip 2 ima povećan rizik da će i sama oboljeti od iste bolesti, ako oba roditelja imaju šećernu bolest tip 2, rizik od oboljevanja iznosi oko 40 %. Do sada je poznato više genetskih promjena koje bi mogle biti povezane s razvojem bolesti. Smatra se da se radi o poligenetskom nasljeđivanju. Genetske promjene koje se povezuju sa šećernom bolesti tip 2 uključuju mutacije različitih gena za različite proteine: transkripcijski faktor 7 (TCF7L2), peroksisom proliferacijski-aktivirajući receptor gama (PPAR-gama), o ATP-u ovisan kalijski kanal β-stanica i druge. Danas se najviše spominje SNP mutacija TCF7L2 gena kao najjači prediktor šećerne bolesti tip 2. Mehanizmi koji povezuju te genetske promjene s razvojem bolesti nisu jasni, ali smatra se da su povezani s razvojem oslabljene funkcije β-stanica Langerhansovih otočića te smanjenim lučenjem inzulina.
Inzulinska rezistencija. Inzulinska rezistencija ključna je u patogenezi šećerne bolesti tip 2. Ona podrazumijeva izostanak učinka inzulina uz očuvanu sekreciju iz β-stanica i normalnu razinu u cirkulaciji. Mehanizam nastanka inzulinske rezistencije nije jasan i vjerojatno je multifaktorske geneze. Rana istraživanja bila su fokusirana samo na poremećaje inzulinskoga receptora, kao što je to slučaj uslijed nekih genetskih sidroma (leprehaunizam, Rabson-Mendenhallov sindrom, inzulinska rezistencija tip A), ali oni se danas opisuju kao posebni, specifični oblici šećerne bolesti, s obzirom na to da je kod njih jasan genetski defekt koji dovodi do poremećaja. Današnja istraživanja usmjerena su na unutarstanične signalne puteve koje se smatra odgovornima za razvoj inzulinske rezistencije. Različiti poremećaji unutarstaničnih signalnih puteva mogu biti odgovorni za izostanak translacije GLUT4 transportnog proteina do stanične membrane kako bi se ostvario ulazak glukoze u stanicu. Moguću ulogu u nastanku inzulinske rezistencije ima i nakupljanje slobodnih masnih kiselina u stanicama jetre i mišića. Njihovi metaboliti dovode do fosforilacije različitih molekula koje sudjeluju u inzulinskom signalnom putu što dovodi do poremećaja u njihovoj funkciji. Debljina se smatra značajnim čimbenikom rizika za nastanak inzulinske rezistencije i to iz dva razloga: centralna ili intraabdominalna pretilost podložna je lipolizi zbog čega se stvaraju slobodne masne kiseline koje se mogu odlagati u stanice mišića i jetre, te zbog same endokrine aktivnosti masnoga tkiva, odnosno lučenja različitih medijatora (adipokina) koji mogu uzrokovati unutarstanične poremećaje inzulinske signalizacije. Tjelesna neaktivnost također je bitan čimbenik inzulinske rezistencije jer smanjena fizička aktivnost dovodi do smanjenoga iskorištavanja slobodnih masnih kiselina i njihova posljedičnog nakupljanja u stanicama.
Sindromi inzulinske rezistencije. Inzulinska rezistencija javlja se i u nekim bolestima i stanjima koje nazivamo sindromima inzulinske rezistencije: metabolički sindrom, inzulinska rezistencija tip A i tip B te sindrom policističnih jajnika. Metabolički sindrom uključuje centralni tip pretilosti, hipertenziju, hiperlipidemiju te inzulinsku rezistenciju i posljedičnu hiperglikemiju, a povezan je s povećanim kardiovaskularnim rizikom. Inzulinska rezistencija tip A javlja se kod mlađih žena te je karakterizirana izraženom hiperinzulinemijom, debljinom i hiperandrogenizmom, što je uzrokovano defektom unutarstaničnoga signalnog puta inzulina. Sindrom inzulinske rezistencija tip B pogađa srednjovječne žene te je karakteriziran hiperizulinemijom, hiperandrogenizmom te pojavom drugih autoimunih bolesti, a uzrokovana je autoantitijelima koja se vežu izravno na inzulinski receptor te ga blokiraju.
Poremećaj inzulinske sekrecije. Početno se inzulinska rezistencija pokušava savladati pojačanim lučenjem inzulina zbog čega nastaje hiperinzulinemija. Hiperinzulinemija može jedno vrijeme kompenzirati inzulinsku rezistenciju, što će se očitovati kao hiperinzulinemija i euglikemija. Postupno se taj kompenzacijski mehanizam gubi i dolazi do smanjenja produkcije inzulina te nastaje relativni nedostatak inzulina. To se tumači iscrpljivanjem i oštećivanjem samih β-stanica uslijed hiperglikemije i stalne hipersekrecije inzulina. Studije na životinjskim modelima objašnjavaju da je ovo oštećenje β-stanica posljedica nakupljanja i precipitacije proteina amilina u Langerhansovim otočićima, a amilin je polipeptid koji se sintetizira u β-stanicama i luči zajedno s inzulinom. Kronična hipersekrecija dovodi do nakupljanja i precipitiranja proteina amilina unutar otočića, što može biti povezano s oštećenjem samih β-stanica i njihovom smanjenom funkcijom. Konačno se i u šećernoj bolesti tip 2 razvija hipoinzulinemija i hiperglikemija. Taj period razvoja promjena je dug, može trajati duži niz godina.
Okolišni čimbenici rizika. Kako je već opisano, debljina je glavni čimbenik rizika za razvoj šećerne bolesti tip 2. Osobe koji imaju indeks tjelesne mase veći od 30 kg/m2 imaju deset puta veći rizik da obole od šećerne bolesti tip 2. Ipak, nemaju sve pretile osobe šećernu bolest tip 2, što ukazuje na postojanje genetske podloge u razvoju bolesti. Debljina, osobito centralni tip, povezana je s razvojem inzulinske rezistencije koja je primarni događaj u patogenezi šećerne bolesti tip 2. Starija životna dob također se smatra čimbenikom rizika u razvoju bolesti. Procjenjuje se da je ovom bolesti zahvaćeno oko 10 % populacije iznad 65 godina života, a da je 70 % svih oboljelih od šećerne bolesti tip 2 u dobi iznad 50 godina života. Čimbenici rizika za razvoj šećerne bolesti prikazani su zbirno u Tablici 8.1.
Metabolički poremećaji. Razvoj metaboličkih poremećaja u šećernoj bolesti tip 2 nešto se razlikuje od tip 1. Razvoj metaboličkih poremećaja sporiji je i dugotrajniji. Kod oboljelih nalazimo relativan manjak inzulina što se očituje količinom nedostatnom za održavanje euglikemije, ali dostatnom da bi se suprimirala lipoliza i proteoliza. Zbog toga je i proizvodnja ketonskih tijela manja i razvoj ketoacidoze rjeđi je nego kod bolesnika sa šećernom bolesti tip 1 gdje nalazimo apsolutni manjak inzulina. Hiperglikemija se kod ovih bolesnika razvija polagano, mjesecima i godinama, a isto tako raste i bubrežni prag za glikozuriju, tako da se i klasični tzv. osmotski simptomi (poliurija, polidipsija) javljaju u blažim oblicima.
|
Tablica 8.1. Čimbenici rizika za razvoj šećerne bolesti tip 2
|
- Obiteljska anamneza pozitivna na šećernu bolest tip 2
- Debljina (indeks tjelesne mase viši od 25 kg/m2)
- Tjelesna neaktivnost
- Rasna/etička pripadnost (Afroamerikanci, Latinoamerikanci)
- Ranije otkrivena IFG ili IGT ili glikozirani hemoglobin 5,7 - 6,4 %
- Gestacijski dijabetes ili rađanje djeteta porođajne mase veće od 4 kg
- Arterijska hipertenzija (arterijski tlak veći od 140/90 mmHg)
- HDL kolesterol manji od 0,9 mmol/L i/ili trigliceridi viši od 2,82 mmol/L
- Sindrom policističnih jajnika ili akantosis nigrikans
- Povijest bolesti na kardiovaskularne bolesti
|
OSTALI, SPECIFIČNI OBLICI ŠEĆERNE BOLESTI
MODY (od engl. maturity-onset diabetes of the young). MODY je rijedak monogenetski poremećaj koji se javlja u kasnoj adolescentnoj ili mlađoj odrasloj dobi. Radi se o monogenetskom poremećaju koji se prenosi autosomno dominantno s penetracijom od 80 %. Pacijenti obično nisu pretili i nemaju karakteristike metaboličkoga sindroma. Hiperglikemija je posljedica oštećene sekrecije inzulina ovisne o glukozi. Ovisno o genetskome defektu, razlikujemo šest oblika MODY-a s različitom učestalosti (Tablica 8.2.). Osim MODY-a tip 2 koji je uzrokovan mutacijom gena za glukokinazu (GCK), ostali su uzrokovani mutacijom gena za nuklearni transkripcijski faktor (HNF, engl. hepatocyte nuclear transcription factor). MODY tip 2 karakterizira smanjeno lučenje bazalnoga inzulina što se očituje povećanom glikemijom natašte, ali ostaje dostatno lučenje inzulina postprandijalno, pa je kod ovih bolesnika glikozilirani hemoglobin obično urednih vrijednosti, a i manja je učestalost razvoja mikrovaskularnih komplikacija te samo liječenje nije uvijek potrebno. MODY se uspješno liječi pravilnom prehranom i oralnim hipoglikemicima kroz duži niz godina, ali s vremenom se mora započeti i liječenje inzulinima. MODY tip 1 i tip 3 dobro reagiraju na sulfonilureju. MODY tip V povezan je i s razvojem renalnih cista.
|
Tablica 8.2. MODY oblici
|
|
Tip
|
Mutacija
|
Učestalost
|
|
MODY tip 1
|
HNF-4α
|
Rijetko
|
|
MODY tip 2
|
Glukokinaza
|
Manje rijetko
|
|
MODY tip 3
|
NHF-1α
|
Najučestalije
|
|
MODY tip 4
|
PDX1
|
Vrlo rijetko
|
|
MODY tip 5
|
HNF-1β
|
Vrlo rijetko
|
|
MODY tip 6
|
Neuro D1
|
Vrlo rijetko
|
LADA (od engl. latent autoimmune diabetes in adults). LADA predstavlja latentni autoimuno uzrokovan dijabetes koji se razvija u odrasloj dobi. Iako kod oboljelih nalazimo autoantitijela na β-stanice, njihova destrukcija je polagana i traje godinama. Među pacijentima koji imaju klinička obilježja šećerne bolesti tip 2, LADA je prisutna kod 10 % starijih od 35 godina, a 25 % kod mlađih od 35 godina. Prospektivne studije pokazale su da bolesnici kod kojih je prisutno više autoantitijela na β-stanice razvijaju njihovu značajnu destrukciju u roku kraćem od pet godina, dok bolesnici koji imaju prisutno samo jedno autoantitijelo (npr. GAD) to razvijaju nakon pet godina. Iako proces propadanja β-stanica koji će dovesti do šećerne bolesti može trajati i do dvanaest godina, oštećenu toleranciju glukoze možemo otkriti i puno ranije odgovarajućim testovima, stoga naziv latentni dijabetes nije u potpunosti ispravan.
Gestacijski dijabetes. Intolerancija glukoze koja se razvije tijekom trudnoće klasificira se kao gestacijski dijabetes. Inzulinska rezistencija povezana je s metaboličkim poremećajima u trudnoći, a povećana potreba za inzulinom može dovesti do razvoja predijabetesa ili dijabetesa. Smanjena osjetljivost stanica na inzulin smatra se posljedicom djelovanja placentalnih hormona. Uslijed toga dolazi do povećane potrebe za sekrecijom inzulina iz β-stanica kao kompenzatornoga mehanizma. Kod genetski predisponiranih žena taj kompenzacijski mehanizam može biti nedostatan te se razvija hiperglikemija. Gestacijski dijabetes razvija se kod 5 do 10 % trudnica. Kod većine žena dolazi do normalizacije glikemije nakon poroda, ali ostaje rizik od razvoja šećerne bolesti u sljedećih deset do dvadeset godina (35 do 60 %).
Predijabetes. Predijabetes je kliničko stanje koje još ne zadovoljava kriterije šećerne bolesti, ali ima tendenciju razvoja svih kriterija. Uključuje termine oštećena tolerancija glukoze (IGT, engl. impaired glucose tolerance) te oštećena glikemija natašte (IFG, engl. impaired fasting glycaemia). Takve osobe imaju 40 % izgleda da u sljedećih pet godina razviju šećernu bolest. Kriteriji za IGT i IFG opisani su u dijagnostici šećerne bolesti.
KLINIČKA SLIKA ŠEĆERNE BOLESTI
Šećerna bolest tip 1 ima produljen asimptomatski period u kojemu dolazi do postupne destrukcije β-stanica. Taj period može trajati i više godina. Kada dođe do kritičnoga smanjenja mase β-stanica, dolazi do razvoja kliničkih manifestacija. Simptomi i znakovi obično se brzo razvijaju (dani, tjedni), a nekada je prvi znak bolesti akutno nastala komplikacija, najčešće dijabetička ketoacidoza. Klasični simptomi podrazumijevaju poliuriju, polidipsiju, polifagiju, poremećaje vida (zamagljen vid), parestezije, umor i malaksalost te gubitak na tjelesnoj težini. Simptomi se razvijaju kroz nekoliko dana ili tjedana kod do tada zdrave djece ili adolescenata. Prijelaz iz asimptomatske u simptomatsku fazu bolesti često je potaknut nekim drugim akutnim oboljenjem koje zahtijeva povećanu potrebu za inzulinom, a najčešće su to akutne infekcije. S obzirom na to da kod ovih bolesnika dolazi do izraženoga deficita inzulina, većina bolesnika zahtijeva visoke doze inzulina kako bi se korigirao metabolički poremećaj koji je nastao. Ponekad nakon uvođenja inzulina u terapiju dolazi do takozvanoga „honeymoon“ perioda, tj. perioda u kojemu bolesnici zahtijevaju manje doze inzulina za normalne metaboličke potrebe, a to je posljedica parcijalnoga oporavka lučenja inzulina iz β-stanica te smanjenja inzulinske rezistencije nakon liječenja bolesti koja je potakla razvoj kliničkih manifestacija. Taj period traje od nekoliko tjedana do nekoliko mjeseci. Konačno ipak dolazi do razvoja apsolutnoga manjka inzulina što zahtijeva odgovarajuću suspstitucijsku terapiju inzulinom.
Šećerna bolest tip 2. Klasični simptomi hiperglikemije (poliurija, polifagija, polidipsija) manje su izraženi kod bolesnika sa šećernom bolesti tip 2, nego kod bolesnika sa šećernom bolesti tip 1. Ovi su bolesnici dugo asimptomatski, a prvi su simptomi nespecifični, na primjer kronični umor i malaksalost. Kod ovih bolesnika nalazimo i učestale infekcije, posebice gljivične infekcije kože te infekcije urinarnoga sustava. Također karakteristično je i sporije cijeljenje rana. Bolest se često dijagnosticira slučajno u asimptomatskoj fazi ili se dijagnosticira nakon razvoja neke od kronične komplikacija. Glavne kliničke razlike između šećerne bolesti tip 1 i 2 dane su u Tablici 8.3.
|
Tablica 8.3. Glavne kliničke razlike između šećerne bolesti tip 1 i 2
|
|
Klinička karakteristika
|
Šećerna bolest tip 1
|
Šećerna bolest tip 2
|
|
Uobičajena dob nastanka bolesti
|
< 30 godina
|
> 50 godina
|
|
Trajanje simptoma
|
Dani, tjedni
|
Mjeseci, godine
|
|
Tjelesna masa
|
Normalna ili smanjena
|
Povećana
|
|
Ketonurija
|
Da
|
Ne
|
|
Rapidno pogoršanje bez inzulina
|
Da
|
Ne
|
|
Nalaz autoantitijela
|
Da
|
Ne
|
|
Komplikacije u vrijeme dijagnoze
|
Ne
|
Često
|
|
Obiteljska anamneza na dijabetes
|
Nije česta
|
Česta
|
|
Druge autoimune bolesti
|
Često
|
Rijetko
|
DIJAGNOZA I OBRADA BOLESNIKA SA ŠEĆERNOM BOLESTI
Dijagnostika šećerne bolesti temelji se na laboratorijskim pretragama krvi i urina. Bolest se često dijagnosticira slučajnom provjerom glikemije kod asimptomatskih bolesnika. Nakon potvrđene dijagnoze potrebno je provesti detaljniju obradu bolesnika s ciljem otkrivanja komplikacija u što ranijim fazama.
Etiološko razlikovanje šećerne bolesti većinom se temelji na osnovi kliničkih karakteristika i vremena pojave bolesti. Osobe sa šećernom bolesti tip 1 imaju sljedeće karakteristike: 1) pojavnost bolesti najčešće prije tridesete godine života; 2) bolesnici su obično mršavi; 3) bolest zahtijeva inzulin kao inicijalnu terapiju; 4) imaju sklonost razvoju ketoacidoze te 5) imaju povećan rizik za obolijevanje od drugih autoimunih bolesti (bolesti štitnjače, adrenalna insuficijencija, perniciozna anemija, vitiligo, celijakija). Osobe sa šećernom bolesti tip 2 imaju sljedeće karakteristike: 1) bolest se najčešće razvija nakon tridesete godine života; 2) bolesnici su obično pretili (80 % bolesnika je pretilo u trenu postavljanja dijagnoze); 3) ne zahtijevaju inicijalnu inzulinsku terapiju te 4) često imaju pridružene karakteristike metaboličkoga sindroma (arterijska hipertenzija, dislipidemija) ili druge bolesti povezane s inzulinskom rezistencijom (npr. sindrom policističnih jajnika).
Standardne laboratorijske pretrage u dijagnostici šećerne bolesti uključuju određivanje koncentracije glukoze u plazmi (GUP), test opterećenja glukozom (eng. oral glucose tolerance test - oGTT) te određivanje glikiranog hemoglobina (HbA1c).
Određivanje koncentracije glukoze u plazmi (GUP) venske krvi najpouzdanija je metoda u dijagnostici šećerne bolesti. Koncentracije glukoze u plazmi više su od vrijednosti u punoj krvi za 10 do 15 % zbog odsutnosti krvnih stanica. Klasično, GUP mjerimo laboratorijski na temelju enzimatske reakcije (glukoza oksidaza) i to predstavlja jeftin, automatiziran i pouzdan postupak, ili putem ručnih glukometara kojima se kolorimetrijskom metodom određuje koncentracije glukoze u kapi kapilarne krvi uzete iz jagodice prsta. Vrijednost GUP-a od 7 mmol/L ili više natašte (odnosno nakon više od osam sati od zadnjega obroka) ili vrijedost GUP-a viša od 11,1 nakon jela u više od jednog mjerenja potvrđuje dijagnozu šećerne bolesti.
Test opterećenja glukozom. Oralni test opterećenja glukozom (oGTT) izvodi se kod bolesnika kojima je vrijednost GUP-a natašte između 6,1 i 7,0 mmol/L ili 7,8 do 11,1 mmol/L postprandijalno. Da bi rezultat testa bio pouzdaniji, preporuča se tri dana prije vršenja ispitivanja osigurati unos ugljikohidrata u minimalnoj dozi od 150 do 200 g na dan, osobito kod bolesnika koji su prethodno bili na dijeti sa smanjenim unosom ugljikohidrata. Na dan testiranja bolesnik ne smije od ponoći uzimati ništa oralno. Test podrazumijeva unos standardizirane otopine 75 g glukoze u 300 ml vode. Pripremljenu otopinu bolesnik treba popiti kroz pet minuta. Krv se uzorkuje u 0. i 120. minuti testiranja kada se određuje GUP. Interpretacija testa prikazana je u Tablici 8.4.
|
Tablica 8.4. Interpretacija oGTT testa
|
|
oGTT
|
Vrijednosti u 0. minuti (mmol/L)
|
Vrijednosti u 120. minuti (mmol/L)
|
|
Oštećena glikemija natašte (IFG)
|
6,1 - 6,9
|
< 7,8
|
|
Oštećena tolerancija glukoze (ITG)
|
< 7,0
|
7,8 - 11,0
|
|
Šećerna bolest
|
≥ 7,0
|
≥ 11,1
|
Glikirani hemoglobin (HbA1c) pokazatelj je dugoročne glikemije kod bolesnika (tjedni, mjeseci) te je danas zlatni standard u dijagnostici šećerne bolestu, a koristan je i u objektiviziranju dugoročne glikemije kod bolesnika s razvijenom šećernom bolesti. Glikacija hemoglobina posljedica je dugotrajne hiperglikemije pri čemu se glukoza kovalentno veže na druge šećere i slobodne amino skupine na α i β-lancu hemoglobina, povećavajući frakciju glikoziliranoga hemoglobina. Glikiranjem se mijenja električni naboj (postaje negativniji) zbog čega se glikozilirani hemoglobin može separirati od neglikoziliranog hemoglobina. Glikirani se hemoglobin može prikazivati kao ukupni glikozilirani hemoglobin (GHb), HbA1 frakcija ili HbA1c frakcija. Većina zemalja glikozilirani hemoglobin prikazuje kao HbA1c frakciju, iako se često koristi i IFCC referentni sustav. Brzina formiranja HbA1c hemoglobina izravno je proporcionalna koncentraciji glukoze u krvi tako da porast od 1 % HbA1c odgovara prosječnom povećanju glukoze u krvi od oko 2 mmol/L. Iako koncentracija HbA1c hemoglobina reflektira prosječnu glikemiju tijekom prosječnoga trajanja života eritrocita (120 dana), polovina eritrocita biva zamijenjena u 60 dana pa je HbA1c najosjetljiviji na promjene u kontroli glikemije koje se događaju u mjesecu prije mjerenja. Vrijednost glikiranoga hemoglobina izračunava se u postocima ( %) ili u mmol/mol, a normalne vrijednosti su 6,5 do 7,5 %, odnosno 48 do 59 mmol/mol. Vrijednosti HbA1c mogu biti niže kod anemičnih bolesnika i u trudnica, a tumačenje nalaza može biti otežano kod bolesnika s uremijom ili hemoglobinopatijama.
Trenutne preporuke ADA-e (engl. American Diabetes Association) za dijagnostiku šećerne bolesti uključuju sljedeće kriterije: a) klasični simptomi hiperglikemije (poliurija, polidipsija, polifagija, neobjašnjen gubitak tjelesne mase) plus slučajno određen GUP iznad 11,1 mmol/L; ili b) GUP natašte (ili nakon 8-satnog gladovanja) 7.0 mmol/L ili viši; ili c) GUP u 120. minuti nakon uzimanja 75 g glukoze (oGTT) 11.1 mmol/L ili viši; ili d) HbA1c ≥ 6.5 %.
Nestandardne laboratorijske pretrage. Među nestandardne pretrage ubrajamo određivanje koncentracije inzulina i C-peptida te određivanje protutijela na β-stanice. Mjerenje serumske koncentracije inzulina ili C-peptida ne mora uvijek biti korisno u razlikovanju šećerne bolesti tip 1 od tip 2, ali niska razina C-peptida u serumu bolesnika potvrđuje potrebu za primjenom inzulina. Mnoge osobe s dijagnosticiranom šećernom bolesti tip 1 još neko vrijeme imaju održanu produkciju C-peptida. Određivanje protutijela na β-stanice korisno je u potvrdi šećerne bolesti tip 1. Najčešće se određuju ICA, GAD65, IAA, IA-2 te ZnT8 protutijela.
OPĆI TERAPIJSKI PRISTUP I DIJETOTERAPIJA
Glavni cilj u liječenju šećerne bolesti je održavati glikemiju urednom jer na taj se način sprečava nastanak brojnih mikro- i makrovaskularnih komplikacija koje prate ovu bolest. ADA (engl. American diabetes association) i EASD (engl. European association for the study of diabetes) preporučavaju snižavanje HbA1c do 7,0 %, a to se može postići održavanjem prosječnoga GUP-a između 8,3 i 8,9 mmol/L.
Ciljne vrijednosti glikemije prema Hrvatskim smjernicama za farmakološko liječenje šećerne bolesti tip 2 iz 2016. godine uključuju nekoliko postulata: 1) kod odraslih se osoba vrijednost HbA1c oko ili ispod 7 % smatra prihvatljivom jer se njome smanjuje učestalost kasnijih komplikacija; 2) kod mlađih osoba, s kraćim trajanjem bolesti, osoba s očekivanim dužim životnim vijekom i osoba bez prisutnosti značajnijih kardiovaskularnih bolesti teži se strožoj kontroli glikemije - njihov HbA1c treba držati između 6,0 i 6,5 %, ako nema sklonosti razvoju hipoglikemije; 3) kod osoba starije životne dobi¸ osoba s prisutnim kardiovaskularnim i drugim komorbiditetima, osoba kod kojih bolest traje duže i osoba sklonijih hipoglikemijama ne preporuča se stroga normoglikemija i kod njih su vrijednosti HbA1c između 7,5 i 8,0 % prihvatljive.
Osnovni terapijski princip liječenja šećerne bolesti je pridržavanje načela pravilne (dijabetičke) prehrane i bavljenje odgovarajućom fizičkom aktivnošću. Ako se glikemija ne može postići promjenom životnoga stila, pristupa se farmakološkom liječenju šećerne bolesti, a ono podrazumijeva primjenu peroralih antidijabetika i/ili primjenu inzulinske terapije.
Dijabetička prehrana. Pravilna prehrana osnovna je i polazišna točka u liječenju šećerne bolesti. Osobe koje se ne pridržavaju te mjere teško da će postići normoglikemiju, bez obzira na farmakološko liječenje. Novije studije pokazuju da sama dijabetička prehrana može pridonijeti regulaciji glikemije i da smanjuje vrijednost glikoziliranoga hemoglobina za 1,0 do 2,0 %. Svi bolesnici s novodijagnosticiranom šećernom bolesti moraju proći prehrambeno savjetovalište kako bi ih se podučilo o potrebama energijskoga unosa, broju obroka, njihovome sastavu, potrebama za pojedinim nutrijentima i drugo. Savjetovališta su uglavnom ustrojena pri centrima koji se bave šećernom bolesti i vode ih obučeni nutricionisti i liječnici.
Dnevne energetske potrebe. Prosječno se bolesnicima koji se ne izlažu većim fizičkim naporima preporuča dnevni energetski unos od 20 do 25 kcal po kilogramu tjelesne mase. Uz takav se pristup često ne postiže učinak mršavljenja pa se, ako je to potrebno, dodatno preporučuje toj vrijednosti oduzeti 500 kcal na dan. Ovisno o tjelesnoj aktivnosti (mlađi i aktivniji bolesnici), energetske potrebe mogu se pojačati s još dodatnih 3 - 10 kcal po kilogramu tjelesne mase.
Broj obroka. Klasično, bolesnici koji se liječe dijetoterapijom i/ili oralnim hipoglikemicima, kao i bolesnici koji su na tzv. bazal-oralnoj (BOT) terapiji, uzimaju pet obroka na dan, podijeljena na tri glavna obroka i dva međuobroka. Bolesnici koji se liječe predmiješanim inzulinima te bolesnici koji su na intenziviranoj (bazal-bolus) inzulinskoj terapiji u pravilu trebaju imati tri glavna obroka na dan, bez međuobroka.
Sastav nutrijenata. Zastupljenost ugljikohidrata trebala bi iznositi 45 – 60 % od ukupne dnevne energetske potrebe. Preporuča se unos složenih ugljikohidrata s niskim glikemijskim indeksom (voće, povrće, integralne žitarice, mlijeko s manjim postotkom mliječne masti). Ne preporuča se unos rafiniranih ugljikohidrata i ugljikohidrata s visokim glikemijskim indeksom. Za slađenje napitaka treba koristiti umjetna sladila. Preporučeni unos proteina trebao bi iznositi 15 - 20 % od ukupnoga dnevnoga energijskog unosa. Kod bolesnika s bubrežnom bolesti unos proteina treba ograničiti na 0,8 g/kg tjelesne mase. Također, pri redukciji tjelesne mase treba izbjegavati dijete s visokim proteinskim udjelom. Udio masti trebao bi iznositi do 35 % od ukupnoga dnevnog energijskog unosa (do 10 % u obliku zasićenih masnih kiselina, a 10 - 20 % u obliku nezasićenih masnih kiselina).
Ostale preporuke. Dopuštene su minimalne količine alkohola, iako ga trebaju izbjegavati bolesnici koji uzimaju inzulin ili oralne hipoglikemike, s obzirom na to da metaboliti alkohola blokiraju glukoneogenezu i mogu pridonijeti razvoju hipoglikemije. Alkohol trebaju uzimati za vrijeme obroka i izbjegavati unos prije spavanja zbog mogućnosti noćnih hipoglikemija. S obzirom na to da je kod ovih bolesnika česta i arterijska hipertezija, preporuča se smanjiti unos soli do 6 g na dan, a kod bolesnika s dijabetičkom nefropatijom i manje. Kod primjene enteralnih pripravaka kao zamjenskih obroka (npr. u jedinicama intenzivnoga liječenja, kod bolesnika s kaheksijom) treba odabrati pripravke prilagođene oboljelima od ove bolesti. Takvi su pripravci niskokalorični, sadrže pretežno sporo otpuštajuće ugljikohidrate niskoga glikemijskog indeksa, nezasićene masne kiseline i prehrambena vlakna.
ORALNI ANTIHIPERGLIKEMICI
Klasično, oralne antihiperglikemike dijelimo u nekoliko skupina prema njihovom načinu djelovanja: 1) lijekovi koji primarno stimuliraju inzulinsku sekreciju vežući se za sulfonilureja receptore na površini β-stanica; 2) lijekovi koji primarno smanjuju koncentraciju glukoze u krvi djelovanjem u jetri, mišićima i masnom tkivu; 3) lijekovi koji imaju učinak na apsorpciju glukoze; 4) lijekovi koji djeluju na inkretinski sustav te 5) inhibitori natrij-glukoza kotransportera 2 ili SGLT2 inhibitori.
LIJEKOVI KOJI PRIMARNO STIMULIRAJU INZULINSKU SEKRECIJU
Preparati sulfonilureje. Primarni mehanizam njihova djelovanja je poticanje sekrecije inzulina iz β-stanica vezanjem na specifične receptore koji se nalaze na površini stanica. Aktivacija receptora za sulfonilureju dovodi do zatvaranja kalijskih kanala i posljedične depolarizacije β-stanica. Stanje depolarizacije omogućava ulazak kalcija u stanice što pogoduje otpuštanju inzulina. Ova skupina lijekova primjenjuje se samo u šećernoj bolesti tip 2 jer zahtijeva postojanje funkcionalnih β-stanica, što nije slučaj u šećernoj bolesti tip 1. Ovi se lijekovi metaboliziraju u jetri, a njihovi su metaboliti slabijega djelovanja, bilo da su aktivni, bilo neaktivni, osim acetoheksamida čiji su metaboliti aktivniji od samoga lijeka. Metaboliti se izlučuju putem bubrega, osim kod preparata sulfonilureje druge generacije koji se dijelom izlučuju i putem žuči, pa su kontraindicirani kod bolesnika s teže oštećenom funkcijom jetre ili bubrega. Najčešće su nuspojave hipoglikemija i dobitak na tjelesnoj masi. Reakcije idiosinkrazije su rijetke, a očituju se kao kožni osipi ili hematološkom toksičnošću (leukopenija, trombocitopenija) s učestalošću od oko 0,1 %. Iako ova skupina lijekova broji četiri generacije, danas se zbog svojih prednosti koriste najčešće gliklazid i glimepirid, odnosno glikvidon.
Gliklazid je preparat sulfonilureje sa srednjim vremenom aktivnosti koje obično iznosi 12 sati, no danas se koriste gliklazidi s produženim otpuštanjem koji se primjenjuju u jednoj dnevnoj dozi, a maksimalna dnevna doza je 120 mg. Lijek se metabolizira u jetri, a nastali metaboliti i konjugati nemaju hipoglikemijski učinak.
Glimepirid je lijek s dužim vremenom djelovanja i može se uzimati jednom na dan. Preporučena početna doza je 1 mg/dan u jednoj jutarnjoj dozi, a preporučena maksimalna doza je 8 mg/dan. Glimepirid se u cijelosti metabolizira u jetri, a nastali metaboliti su neaktivni i nemaju hipoglikemijski učinak.
Analozi meglitinida. Najpoznatiji analog meglitinida je repaglinid. Taj je lijek strukturno sličan gliburidu (preparat sulfonilureje prve generacije), ali mu nedostaje sulfonilurejski pol. Djeluje tako da se veže na receptore za sulfonilureju što dovodi do zatvaranja kalijskih kanala osjetljivih na ATP. Brzo se apsorbira iz crijeva te se u jetri u potpunosti metabolizira u inaktivne bilijarne produkte. Poluvijek života u plazmi manji je od jednoga sata pa dovodi do pulsnoga lučenja inzulina. Početna doza je 0,5 mg tri puta na dan, 15 minuta prije glavnih obroka. Maksimalna dnevna doza je 16 mg. Kao i ostali preprati sulfonilureje, repaglinid se može kombinirati s metforminom. Glavna nuspojava je hipoglikemija, a kao i ostali preparati sulfonilureje, može izazvati povećanje tjelesne mase. Metabolizira se u jetri preko enzima citokrom P450 pa uzimanje lijekova koji dovode do indukcije ili inhibicije ovih enzima mogu pojačati ili smanjiti metabolizam repaglinida. Ovaj lijek može biti koristan kod starijih osoba ili osoba s oštećenom bubrežnom funkcijom.
Derivati D-fenilalanina. Najpoznatiji lijek iz ove skupine je nateglinid. Taj lijek svoj učinak ostvaruje vežući se za receptore za sulfonilureju što dovodi do zatvaranja kalijskih kanala. Brzo se apsorbira iz crijeva, a vršne vrijednosti u plazmi doseže unutar jednoga sata. Metabolizira se u jetri, a poluvijek života u plazmi mu je sat i pol. Kao i repaglinid, uzrokuje pulsno lučenje inzulina i kada se uzima uoči glavnih obroka smanjuje postprandijalnu hiperglikemiju. Kod većine je bolesnika preporučena doza lijeka 120 mg tri puta na dan pred glavne obroke. Kod bolesnika sa srednje povišenim HbA1C može se davati i u dozi od 60 mg tri puta na dan. Glavne nuspojave su hipoglikemija i dobivanje na tjelesnoj masi.
LIJEKOVI KOJI PRIMANO SMANJUJU KONECNTRACIJU GLUKOZE U KRVI
Metformin. Metformin predstavlja prvu liniju liječenja kod bolesnika sa šećernom bolesti tip 2. Terapijski učinak postiže primarno djelujući na jetru i to tako da povećava aktivnost ATP-aktivirajuće protein kinaze koja smanjuje glukoneogenezu i lipogenezu. Poluvijek života metformina u plazmi iznosi 1,5 do 3 sata, ne veže se za proteine plazme, ne prolazi metabolizam u jetri, a luči se u nepromijenjenom obliku putem bubrega. Metformin poboljšava glikemiju natašte i postprandijalno, kao i hipertrigliceridemiju u pretilih osoba. Također, metformin ne dovodi do dobitka na tjelesnoj masi, što se viđa kod bolesnika koji uzimaju preparate sulfonilureje ili su na inzulinskoj terapiji. Ovaj lijek treba davati s oprezom kod bolesnika s kroničnom bolesti bubrega i smanjenom glomerularnom filtracijom (glomerularna filtracija manja od 20 ml/min predstavlja kontraindikaciju ta primjenom ovog lijeka) jer smanjeno izlučivanje metformina dovodi do porasta njegove koncentracije u krvi i tkivima, što može izazvati prekomjerno stvaranje laktata (moguća pojava laktacidoze). Kod bolesnika s GF-om između 20 i 60 m metformin se ne bi trebao davati, niti bolesnicima s jetrenom insuficijencijom niti bolesnicima koji prekomjerno uzimaju alkohol, zbog sklonosti razvoju laktacidoze (za vrijeme terapije metforminom pojačava se proizvodnja laktata u crijevima i drugim tkivima, a smanjenjem aktivnosti hepatocita ili uslijed alkoholom inducirane redukcije nukleotida smanjuje se klirens laktata). Preporučena dnevna doza metformina je 2000 mg. Obično se počinje manjim dozama podijeljenim u nekoliko dnevnih doza (uz glavne obroke). Najčešće se započinje s dozom od 500 mg dva do tri puta na dan uz glavne obroke. Također, dobar se odgovor postiže i dozom od 850 mg ili 1000 mg dva puta na dan (prije doručka i prije večere). Na raspolaganju su i preparati metformina s produženim otpuštanjem koji se primjenjuju u jednoj dnevnoj dozi, bez potrebe za postupnim titriranjem doze. Najčešće nuspojave metformina su gastrointestinalne: gubitak teka, mučnina, povraćanje, proljev te bol u trbuhu, a javljaju se kod manje od 20 % bolesnika. Te su nuspojave uglavnom ovisne o dozi. Kod 3 do 5 % bolesnika mora se prestati s terapijom metforminom zbog upornih proljeva. U terapijskim dozama nisu zabilježene hipoglikemije zbog čega se metformin obično naziva euglikemijski ili antihiperglikemijski lijek. Kožne (osip) ili hematološke (trombocitopenija, leukopenija) nuspojave su rijetke. Metformin interferira s apsorpcijom, ovisnoj o kalciju, kompleksa vitamina B12-unutarnji faktor u terminalnom ileumu, zbog čega se može razviti deficijencija vitamina B12 nakon dužega perioda uzimanja (godine). Preporuča se periodično određivanje koncentracije vitamina B12, osobito kod bolesnika koji su liječenih metforminom, a imaju razvijenu makrocitnu anemiju ili perifernu neuropatiju. Navedeni se poremećaj može prevenirati pojačanim unosom kalcija. Metformin u terapijskoj dozi smanjuje klirens laktata putem jetre, ali serumske koncentracije laktata minimalno rastu, sve dok ih drugi organi, kao što je bubreg, uspijevaju ukloniti. Ipak, kod bolesnika liječenih metforminom tkivna hipoksija brže dovodi do razvoja hiperlaktatemije i laktacidoze. Ako je bubrežna funkcija oštećena, laktacidoza se može razviti i bez pojačanoga stvaranja laktata i uz terapijske doze metformina.
Tiazolidini. Ovi lijekovi djeluju tako da povećavaju osjetljivost perifernih tkiva na inzulin. Vežu se na nuklearni receptor PPAR-ϒ (peroksisom proliferirajući-aktivirajući aktivator gama) koji dovodi do ekspresije različitih gena uključenih u inzulinski signalizacijski put. Najpoznatiji tiazolidini su roziglitazon i pioglitazon, a oba su učinkovita, bilo kao monoterapija, bilo u kombinaciji sa sulfonilurejom, metforminom ili inzulinom, smanjujući HbA1c za 1 do 2 %. Roziglitazon je povučen s europskoga tržišta nakon 2010. godine kada je na temelju jedne meta-analize utvrđen povećan rizik od nastanka srčanoga udara kod bolesnika koji su uzimali ovaj lijek. Pioglitazon osim na smanjenje koncentracije glukoze u plazmi ima i neke druge učinike kao što su snižavanje koncentracije triglicerida i povišenje HDL kolesterola, kao i povoljan učinak u liječenju nealkoholne masne bolesti jetre. Prepručena dnevna doza pioglitazona iznosi 15 do 45 mg. Najčešće nuspojave su periferni edemi, makularni edem, a neke studije ukazuju i na veći rizik od razvoja karcinoma mokraćnoga mjehura kod uzimanja piglitazona u većim dozama kroz dulje vrijeme.
LIJEKOVI KOJI IMAJU UČINAK NA APSORPCIJU GLUKOZE
Akarboza je inhibitor α-glukozidaze, enzima koji razgrađuje škrob i saharozu na njihove sastavne dijelove. Preporučena početna doza je 50 mg dva puta na dan, a može se povisiti do 100 mg tri puta na dan. Za najbolji učinak na smanjenje postprandijalne glikemije lijek se treba uzimati s prvim zalogajem obroka. Kod 20 - 30 % bolesnika razvija se nuspojava u vidu flatulencije zbog slabije probave ugljikohidrata koji potom pristižu u distalnije dijelove crijeva gdje bakterijska flora dovodi do stvaranja plinova. Kod 3 % bolesnika razvijaju se i uporni proljevi. Ako se akarboza primjenjuje samostalno, nema rizika od razvoja hipoglikemije, no ako se koristi uz sulfonilureju ili inzuline, povećava rizik od nastanka hipoglikemije. Kod nekih se bolesnika može bilježiti lagan porast aminotransferaza, a te se vrijednosti vraćaju u normalu po prestanku uzimanja lijeka.
LIJEKOVI KOJI DJELUJU NA INKRETINSKI SUSTAV
Inkretinski učinak. Glukoza unesena peroralnim putem izaziva trostruko ili četverostruko veći inzulinski odgovor u odnosu na istu količinu glukoze unesenu intravenski. Objašnjenje toga leži u činjenici da peroralno unesena glukoza dovodi do oslobađanja hormona crijeva koje nazivamo inkretini (peptid 1 sličan glukagonu ili GLP-1 te inzulotropni polipeptid ovisan o glukozi ili GIP-1), a cijeli taj mehanizam naziva se inkretinski učinak. Inkretinski učinak smanjen je kod bolesnika sa šećernom bolesti tip 2. Osim učinka na sekreciju inzulina, GLP-11 ima i druge učinke: suprimira sekreciju glukagona, usporava pražnjenje želuca, a u središnjem živčanom sustavu izaziva osjet sitosti. Bolesnici koji uzimaju GLP-1 agoniste imaju smanjen apetit, što je posljedica centralnoga učinka GLP-1 na osjet sitosti i usporenoga pražnjenja želuca
Agonisti GLP-1 receptora. Budući da se GLP-1 brzo razgrađuje djelovanjem enzima DPP-4 (dipeptidil peptidaza 4) i budući da se brzo izlučuje bubregom, poluvijek života GLP-1 iznosi 1 do 2 minute. Zbog toga se nativni GLP-1 ne može primjenjivati u terapijske svrhe, nego su razvijeni njegovi metabolički stabilni analozi, kao što su eksenatid, liraglutid, albiglutid, dulaglutid, liksisenatid i semaglutid (Tablica 8.5.).
|
Tablica 8.5. Osnovne karakteristike agonista GLP1 receptora
|
|
Lijek
|
Poluvijek života
|
Učinak
|
Doziranje
|
|
Eksenatid
|
2,4 sata
|
↓ HbA1c 0,4 %
↓ TM 1,5 - 3 kg
|
Početno 2 x 5 mg sc 60 min prije doručka i večere;
Nakon mjesec dana povisiti dozu na 2 x 10 mcg
|
|
Liraglutid
|
12 sati
|
↓ HbA1c 1 %
↓ TT 1,5 - 3 kg
|
Početno 0,6 mg jednom na dan
Nakon tjedan dana povisiti dozu na 1,2 mg jednom na dan
|
|
Albiglutid
|
5 dana
|
↓ HbA1c 0,8 %
↓ TT 0,5 kg
|
Početno 1 x 30 mg sc tjedno
Postupno povišenje do doze od 1 x 50 mg sc tjedno
|
|
Dulaglutid
|
5 dana
|
↓ HbA1c 1,3 %
↓ TT 0,5 - 3 kg
|
Početno 1 x 0,75 mg sc tjedno
Postupno povišenje do doze od 1,5 mg sc tjedno
|
|
Liksisenatid
|
3 sata
|
↓ HbA1c 0,5 %
↓ TT 1 - 3 kg
|
Početno 1 x 10 mcg prije doručka
Postupno povišenje do doze od 1 x 20 mg sc prije doručka
|
|
Semaglutid
|
7 dana
|
↓ HbA1c 1,8 %
↓ TT 0,5 kg
|
Početno 1 x 0,25 mg sc tjedno
Nakon mjesec dana povisiti dozu na 1 x 0,5 mg sc tjedno
|
Najčešće nuspojave GLP-1 agonista su mučnina (11 - 40 %), povraćanje (4 - 13 %) te proljev (9 - 17 %). Nuspojave su češće kod viših doza. Neke su studije ukazale na moguću povezanost primjene agonista GLP-1 i pojave akutnoga pankreatitisa, stoga se preporuča oprez kod primjene ovih lijekova kod osoba koje su preboljele pankreatitis. Također, pri primjeni eksenatida zabilježeno je pogoršanje bubrežne funkcije, a kod nekih i pojava akutnoga bubrežnog oštećenja. S obzirom na to da je na životinjskim modelima (štakori) zabilježeno da GLP-1 agonisti mogu izazvati medularni karcinom (C-stanice imaju GLP-1 receptore na svojoj površini), ne bi ih se smjelo davati osobama koje imaju pozitivnu anamnezu na karcinom štitne žlijezde.
DPP-4 inhibitori. DPP-4 inhibitori su lijekovi koji inhibiraju DPP-4 enzim i tako produljuju život endogeno stvorenoga GLP-1 i GIP-1. U kliničkoj praksi su: sitagliptin, saksagliptin, alogliptin, linagliptin te vildagliptin.
Sitagliptin u kliničkim studijama pokazuje poboljšanje HbA1c za 0,5 do 1,4 %, bilo da se koristi kao monoterapija, bilo kao dodatak postojećoj terapiji. Uobičajena je doza 100 mg jednom na dan uz prilagođavanje ako se radi o oštećenoj funkciji bubrega (ako je klirens kreatinina između 30 i 50 ml/min preporučena doza je 50 mg, a ako je ispod 30 ml/min 25 mg). Sitagliptin ima neutralan učinak na tjelesnu masu. Glavne su nuspojave, iako rijetke, infekcije gornjih dišnih puteva, a rjeđe se nađu neutropenija, alergijske reakcije te pankreatitis. S obzirom na to da DPP-4 enzimi imaju više supstrata (neuropeptidi, čimbenici rasta, kemokini), njegova supresija nije potpuno istražena u odnosu na konačni klinički učinak.
Saksagliptin pokazuje poboljšanje HbA1c za 0,7 do 0,9 % kada se dodaje drugim lijekovima. Uobičajena doza je 2,5 do 5 mg jednom na dan. Doza od 2,5 mg preporuča se bolesnicima koji imaju klirens kreatinina ispod 50 ml/min. Ne dovodi do smanjenja ni povećanja tjelesne težine. Glavne nuspojave su infekcije gornjega dišnog trakta, glavobolja te urinarne infekcije. Reakcije preosjetljivosti kao urtikarija ili angioedem pojave se kod oko 1,5 % bolesnika u usporedbi s 0,4 % kod placebo bolesnika. Saksagliptin se metabolizira u jetri putem CYP3A4/5 izoenzima te lijekovi koji induciraju ili reduciraju njihovu aktivnost mogu utjecati na farnakokinetiku saksagliptina. Studije su pokazale da taj lijek može pogoršati srčano popuštanje, ali se to bilježi kod bolesnika koji su i ranije imali srčano popuštanje ili su imali povišen NT-proBNP.
Alogliptin poboljšava HbA1c za 0,5 do 0,6 % kada se doda ranijoj terapiji (metformin, sulfonilureja, pioglitazon). Uobičajena doza je 25 mg jednom dnevno. Dozu treba prilagoditi bubrežnoj funkciji: ako je klirens kreatinina 30 do 60 ml/min doza treba biti 12,5 mg, a ako je manji od 30 ml/min, onda 6,25 mg. Glavne nuspojave su pankreatitis, reakcije preosjetljivosti, jetrena insuficijencija te povećani rizik od nastanka srčanoga popuštanja.
Linagliptin poboljšava HbA1c za 0,4 do 0,6 % ako se doda metrominu, sulfonilureji ili pioglitazonu. Preporučena doza je 5 mg na dan. S obzirom na to da se većina lijeka izluči metabolički nepromijenjena putem žuči, nije potrebno posebno doziranje u slučaju oštećene bubrežne funkcije. Glavne nuspojave su infekcije gornjega dišnog sustava te reakcije preosjetljivosti. Pankreatitis je rijetka komplikacija.
Vildagliptin kao dodatna terapija snižava HbA1c za 0,5 do 1 %. Preporučena doza je 50 mg dva puta na dan. Glavne nuspojave su infekcije gornjega respiratornog trakta i glavobolja, a jetrena insuficijencija je rijetka.
INHIBITORI NATRIJ-GLUKOZA KOTRANSPORTERA 2 (SGLT2 INHIBITORI)
Glukoza se slobodno filtrira u glomerulu te se reapsorbira u proksimalnom tubulu pomoću natrij-glukoza kotransportera (SGLT). Smatra se da je SGLT2 odgovoran za 90 % reapsorpcije glukoze u proksimalnom tubulu te njegova inhibicija dovodi do pojačanoga lučenja glukoze urinom (glikozurija) uz smanjenje glukoze u plazmi. Najznačajniji lijekovi iz ove skupine su kanagliflozin, dapagliflozin i empagliflozin.
Kanagliflozin smanjuje prag glikozurije s 10 mmol/L na 4 do 5 mmol/L te reducira HbA1c za 0,6 do 1,0 %, bilo da se koristi sam, bilo da se dodaje drugom lijeku ili inzulinu. Bilježi se i smanjenje tjelesne mase za oko 2 do 5 kg. Uobičajena doza je 100 mg jednom dnevno, a kod bolesnika s očuvanom bubrežnom funkcijom može se povisiti i na najviše 300 mg na dan.
Dapagliflozin reducira HbA1c za 0,5 do 0,8 % kao monoterapija ili kao dodatak terapiji od ranije. Dovodi i do gubitka na tjelesnoj težini od 2 do 4 kg. Uobičajena je doza 10 mg na dan, ali se kod bolesnika s oštećenom jetrenom funkcijom preporuča doza od 5 mg/dan. U Hrvatskoj nema registiranoga čistog dapagliflozina, nego njegova fiksna kombinacija s metforminom.
Empagliflozin reducira HbA1c za 0,5 do 0,7 % kao monoterapija ili kao dodatak terapiji od ranije. Dovodi i do gubitka na tjelesnoj težini od 2 do 3 kg. Uobičajena doza je 10 mg na dan, a može se povisiti do 25 mg na dan.
Učinkovitost SGLT2 inhibitora smanjena je u kroničnoj bubrežnoj bolesti, a treba imati na umu da ovi lijekovi mogu dovesti i do povišenja kreatinina i smanjenja glomerularne filtracije. Kanagliflozin je kontraindiciran ako je glomerularna filtracija manja od 45 ml/min/1,73m2. Najčešće nuspojave primjene ovih lijekova su infekcije mokraćnoga sustava, osobito gljivične. Glikozurija može dovesti i do smanjenja intravaskularnoga volumena te hipotenzije. U terapiji kanagliflozinom primjećena je i češća pojava fraktura gornjih udova (upitno je radi li se o posljedici djelovanja na kost ili hipotenziji). Također, bilježi se i blagi porast LDL kolesterola (3 – 8 %).
INZULINSKA TERAPIJA
Inzulin je indiciran u liječenju šećerne bolesti tip 1, kao i kod bolesnika sa šećernom bolesti tip 2 i inzulinopenijom kod kojih hiperglikemija ne reagira na dijetoterapiju i terapiju oralnim antihiperglikemicima.
Karakteristike dostupnih inzulinskih preparata. Inzulinski pripravci proizvode se DNA-tehnologijom čime se dobivaju rekombinantne humane strukture. Komercijalni inzulinski preparati međusobno se razlikuju prema vremenu početka i trajanju učinka. Humani inzulin dolazi u dvije formulacije: kao regularni inzulin i kao NPH inzulin (engl. neutral protamine hagedron). Razlikujemo i šest inzulinskih analoga, od toga su tri analoga brzodjelujuća (inzulin lispro, inzulin aspart, inzulin glulizin), a tri su dugodjelujuća (inzulin glargin, inzulin detemir, inzulin degludek). Animalni inzulini više se ne primjenjuju. Svi danas dostupni inzulini sadrže manje od 10 ppm proinzulina te se nazivaju pročišćenima. Pročišćeni inzulinski preprati čuvaju inzulinski potencijal, stoga se držanje na hladnom (u hladnjaku) preporučuje, ali nije od presudne važnosti. Ako se inzulini tijekom putovanja štite od ekstrema hladnoće i vrućine, mogu imati normalan učinak tjednima. Dostupni su u koncentraciji od 100 jed dispenzirano u 10 mL ampule ili u posebnim jednokratnim penovima.
Podjela inzulinskih preparata. Okvirno, inzuline dijelimo u četiri skupine: brzodjelujući inzulini, srednjedugodjelujući inzulini, dugodjelujući inzulini i predmiješani inzulini (Slika 8.1.). U brzodjelujuće inzuline ubrajamo regularni (humani) inzulin i brzodjelujuće inzulinske analoge. Oni dolaze kao čiste otopine neutralnog pH koje sadrže male količine cinka kako bi se poboljšala njihova stabilnost i dužina trajanja. U srednjedugodjelujuće inzuline ubrajamo NPH inzuline. Oni dolaze kao kao mutna suspenzija neutralnoga pH s protamin fosfatnim puferom, a primjenjuju se kao bazalni inzulini u dvije dnevne doze. Dugodjelujući inzulini dolaze kao inzulinski analozi, a danas su na raspolaganju inzulin detemir, glargin i degludec. Predmiješani inzulini kombinacija su kratkodjelujućih inzulinska i NPH inzulina u različitim omjerima. Brzodjelujući i dugodjelujući inzulinski analozi predviđeni su za supkutanu primjenu, dok se regularni inzulin može dati i intravenski. Brzodjelujući inzulini nazivaju se još i prandijalni inzulini jer se njihovom primjenom osigurava dovoljna količina inzulina za obrok, a primjenjuju se u terapijskoj shemi zajedno sa srednjedugodjelujućim ili dugodjelujućim inzulinima. Srednjedugodjelujući i dugodjelujući inzulini nazivaju se i bazalni inzulini jer se njima pokrivaju bazalne potrebe za inzulinom između obroka.
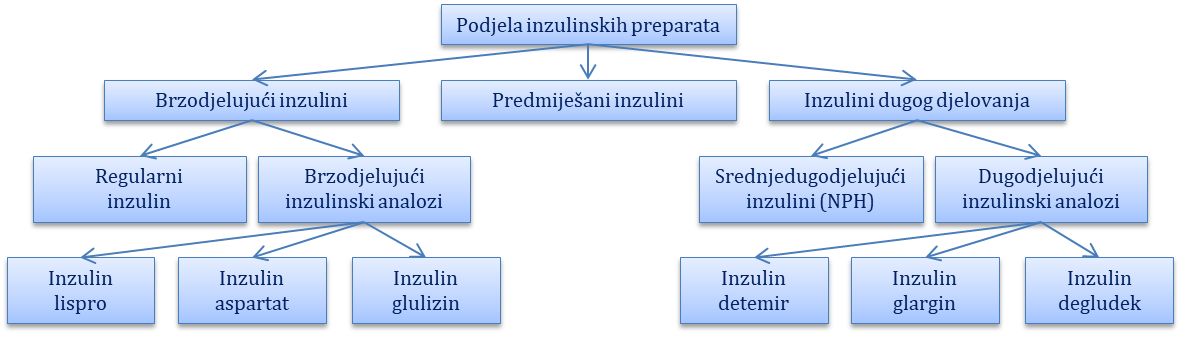
Slika 8.1. Podjela inzulinskih preparata
Regularni (humani) inzulin. Regularni inzulin je inzulin kratkoga djelovanja koji dolazi u čistoj (kristalnoj) otopini obogaćenoj cinkom. Početak djelovanja očekuje se nakon 30 minuta od supkutane primjene i traje 5 do 7 sati. Regularni inzulin može se davati i intravenski kada je to potrebno, npr. u liječenju ketoacidoze. Indiciran je u stanjima kada je potrebna učestala primjena inzulina (poslijeoperacijski, infekcije).
Brzodjelujući inzulinski analozi. U ovu skupinu ubrajamo tri inzulinska peparata: inzulin lispro, inzulin aspartat i inzulin glulizin. Inzulin lispro je inzulinski analog kod kojega je prolin na poziciji B28 zamijenjen lizinom na poziciji B29. Inzulin aspartat je inzulinski analog kojemu je prolin zamijenjen aspartatom na poziciji B28. Inzulin glulizin je inzulinski analog s dvostrukom zamjenom: aspargin na poziciji B3 zamijenjen je lizinom te je lizin na poziciji B29 zamijenjen glutaminom. Uslijed tih izmjena ovi inzulinski analozi imaju manju tendenciju stvaranja konformacije heksamera te zbog toga nakon supkutane primjene brzo disociraju u monomere i brže se apsorbiraju, za razliku od humanoga inzulina koji se zbog heksamerske konformacije sporije disocira u monomere i sporije apsorbira. Navedene izmjene u slijedu aminokiselina ne interferiraju s nekim bazičnim osobitostima (sposobnost vezanja na receptore, poluvijek života, imunogenetika). Optimalno vrijeme za primjenu brzodjelujućih inzulinskih analoga je neposredno prije obroka (ev. 5 - 10 minuta prije obroka, iako se u nekim slučajevima mogu davati i neposredno nakon obroka). Optimalno vrijeme za primjenu regularnog inzulina je 30 minuta prije obroka. Drugi poželjan učinak brozodjelujućih inzulinskih analoga u odnosu na regularni inzulin, vrijeme je učinka od četiri sata bez obzira na dozu, dok je vrijeme djelovanja regularnoga inzulina ovisno o dozi (što je viša doza, duži je i učinak). Brzodjelujući inzulinski analozi koriste se u inzulinskim pumpama.
NPH inzulin. NPH inzulin (neutral protamin Hagedorn, izofan) je humani inzulin srednjedugoga djelovanja što je osigurano različitim izmjenama molekularnoga ustroja. Početak djelovanja NPH inzulina nastupa za 2 do 4 sata s vršnim djelovanjem nakon oko 6 do 7 sati. Budući da je vrijeme djelovanja kraće od 24 sata (u rasponu 10 do 20 sati), većina bolesnika zahtijeva primjenu NPH inzulina u dvije dnevne doze.
Inzulin glargin U100. Inzulin glargin U100 je inzulinski analog dugoga djelovanja kojemu je asparagin na poziciji 21 A lanca zamijenjen glicinom te su mu dodane dvije molekule arginina na karboksi terminalnu skupinu B lanca. Dodani arginini održavaju izolelektričnu točku molekule inzulina bliže neutralnoj, što omogućava bolju topljivost u kiselome okruženju (u odnosu na to, humani inzulin ima izoelektričku točku na pH 5,4). Inzulin glargin čisti je inzulin koji, kada se injicira u neutralni pH medij kakav je u supkutanome masnom tkivu, stvara mikropercipitate koji se postupno otpuštaju u cirkulaciju. Početak djelovanja nastupa nakon sat i pol, postupno se otpušta u cirkulaciju bez stvaranja maksimuma djelovanja (nema vršne koncentracije, koncentracijska krivulja je ravna), a djelovanje se vidi kroz 24 sata. Iz tog se razloga taj inzulin najčešće primjenjuje u jednoj dnevnoj dozi te se tako osigurava inzulin za pokrivanje bazalne glikemije. Inzulin glargin U100 se ne može miješati s ostalim humanim inzulinima jer zahtijeva kiseli pH. Kada se inzulin glargin daje kod bolesnika sa šećernom bolesti tip 1 kao jedna doza prije spavanja, postižu se bolje vrijednosti prandijalne glikemije u odnosu na primjenu NPH inzulina, s manjom učestalošću noćnih hipoglikemija.
Inzulin detemir. Inzulin detemir dugodjelujući je inzulinski analog kojemu je tirozin uklonjen s pozicije B30, a na lizin na poziciji B29 dodan je 14-C lanac masne kiseline (tetradekanska kiselina). Njegov produljeni učinak posljedica je raspadanja heksamera te vezanja za albumine na mjestu aplikacije, kao i u plazmi. Vrijeme djelovanja inzulina detemira je 17 sati. Preporučuje se njegova primjena u jednoj ili dvije doze na dan kako bi se osigurala potrebna bazalna doza inzulina. Dozvoljena je njegova upotreba za vrijeme trudnoće.
Inzulin degludek. Inzulin degludek dugodjelujući je inzulinski analog koji nakon supkutane primjene formira topljive i stabilne multiheksamere koji tvore depo inzulina i postupno se otpuštaju. Tako se omogućava postupno otpuštanje lijeka (odvajanje monomera od multiheksamera) tijekom 42 sata. Ipak, nakon postignute dinamičke ravnoteže, usprkos dugom poluvijeku života, liječenje njegovom primjenom jednom na dan neće dovesti do nakupljanja inzulina u cirkulaciji. Ta svojstva omogućuju veliku fleksibilnost u vremenu primjene inzulina. Studije nisu pokazale značajniju superiornost degludeka naspram glargina U100. Ipak, terapija degludekom povezana je s manjom učestalošću hipoglikemija u odnosu na terapiju glarginom, posebice noćnih hipoglikemija.
Inzulin glargin U300 zajedno s inzulinom degludekom predstavlja bazalni inzulinski analog druge generacije koji za razliku od inzulina glargina U100 ima dugotrajnije otpuštanje (posljedica smanjenja volumena injekcije, a time i površine precipitata), pa je i učinak dulji za oko 5 sati u odnosu na inzulin glargin U100. Studije nisu pokazale bolju kontrolu glikemije, kao ni smanjen rizik od hipoglikemije u odnosu na inzulin glargin U100.
Predmiješani inzulini. Predmiješani inzulinski pripravci predstavljaju mješavine brzodjelujućega inzulina (humani inzulin ili inzulinski analog) i čistoga inzulina kristaliziranoga s protaminom čime se dobije komponenta srednjedugodjelujućega inzulina. Te se mješavine pojavljuju u različitim omjerima: 25/75, 30/70 ili 50/50 (prvi broj označava udio brzodjelujućega inzulina). Takvi se inzulini primjenjuju prije obroka, a pokrivaju i bazalne i prandijalne potrebe za inzulinom. Povezani su s povećanjem tjelesne mase. Novije formulacije predmiješanih inzulina uključuju mješavinju inzulina degludeka i brzodjelujućega inzulina aspartata.
Načini primjene inzulina. Inzulin se može primjenjivati na nekoliko načina: putem igli i štrcaljki (danas rijetko), putem posebnih injektora u obliku olovke (danas najpopularnije) ili inzulinskih pumpi (najčešće namijenjeno mlađoj populaciji). Inzulin se primjenjuje u kroničnoj terapiji supkutano, najčešće u području abdomena, ali može se primijeniti i u području nadlaktica i natkoljenica. Preporuča se izmjena mjesta aplikacije inzulina kako se ne bi razvila fibroza ili lipodistrofija. U hitnim se stanjima određeni inzulini mogu primijeniti i intravenski. Sve se više opisuje i usavršava inhalacijska primjena inzulina.
Nuspojave inzulinske terapije. Najčešće nuspojave povezane s primjenom inzulina su hipoglikemija i porast tjelesne mase, dok su alergija na inzulin, lipodistrofija te imunološki posredovana rezistencija na inzulin rijetke nuspojave.
Hipoglikemija je najčešća nuspojava inzulinske terapije, a posljedica je nesrazmjera između primijenjenoga inzulina i glukoze u cirkulaciji (primjena veće količine inzulina ili uzimanje manjega obroka). Simptomi se dijele u dvije skupine: simptomi autonomnoga živčanog sustava te neuroglikopenični simptomi. Pri koncentraciji glukoze od 3 mmol/L javljaju se simptomi autonomnoga živčanog sustava, i to simpatikusa (tahikardija, palpitacije, preznojavanje, tremor) te parasimpatikusa (glad, povraćanje). Ako se ne reagira na te simptome, dolazi do daljnjega snižavanja koncentracije glukoze, a simptomi manjka glukoze za potrebe funkcioniranja živčanog sustava (neuroglikopenija) javljaju se pri 2,8 mmol/L. Simptomi uključuju gubitak koncentracije, letargiju, konfuziju, glavobolju, smetnje vida, otežan govor, a na kraju dolazi do poremećaja svijesti – kome. Kod opetovanih hipoglikemija dolazi do adaptacije autonomnoga živčanog sustava, što znači da navedeni simptomi mogu izostati. Također, simptomatologija može biti oslabljena i kod bolesnika koji u terapiji imaju beta blokatore (kod njih ostaje znojenje kao dominantan znak). Kod bolesnika s učestalim hipoglikemijama dokazan je brži razvoj klasičnih komplikacija šećerne bolesti. Glavna mjera u rješavanju hipoglikemija povezanih s inzulinskom terapijom je edukacija bolesnika o pravilnoj primjeni inzulina i pravilnoj prehrani. Također ih je potrebno educirati o većoj učestalosti razvoja hipoglikemije uslijed uzimanja alkohola, osobito na prazan želudac te nakon izlaganja fizičkoj aktivnosti. Kod pojave simptoma hipoglikemije bolesnici bi trebali izmjeriti GUP te uzeti 15 g ugljikohidrata (kocka šećera, čokolada i sl.) te nakon 15 minuta ponovno premjeriti GUP, te u slučaju i dalje prisutnih sniženih vrijednosti ponoviti uzimanje ugljikohidrata. Kod teže izraženih hipoglikemija primjenjuje se intravenozna infuzija 40 %-tne glukoze ili se primjenjuje 1 mg glukagona intramuskularno.
Alergija na inzulin rijetka je nuspojava inzulinske terapije. Radi se o klasičnoj IgE posredovanoj osjetljivosti, a simptomi su posljedica degranulacije histamina iz mastocita. Najčešće se radi o lokalnoj reakciji (eritem), ali moguć je i razvoj generalizirane urtikarije. Pojava anafilaksije je rijetka. Kod takvih se bolesnika moraju primjenjivati antihistaminici i kortikosteroidi, a nekada u obzir dolazi i hiposenzibilizacija.
Lipodistrofija označava atrofiju supkutanoga masnog tkiva na mjestu učestaloga injiciranja inzulina, a posljedica je imunološke reakcije. S obzirom na to da su današnji inzulinski preparati pročišćeni, učestalost lipodistrofije je daleko rjeđa. Lipodistrofija se prevenira izmjenom mjesta aplikacije inzulina.
Imunološki posredovana rezistencija na inzulin rjeđa je pojava otkako se ne koriste animalni inzulini. Podrazumijeva pojavu niskog titra IgG antitijela u cirkulaciji koji neutraliziraju učinak inzulina. Klinički se očituje povećanom potrebom za inzulinom da bi se održala normoglikemija.
Strategije inzulinske terapije. Okvirno rečeno, razlikuju se četiri strategije primjene inzulina, a to su: 1) u kombinaciji s oralnim antihiperglikemicima (tzv. bazal-oral terapijska shema – BOT); 2) bazal-plus shema; 3) bazal-bolus shema i 4) liječenje predmiješanim inzulinima. Sve četiri strategije prihvatljive su u liječenju šećerne bolesti tip 2, dok se u liječenju šećerne bolesti tip 1 primjenjuje samo bazal-bolus shema.
Bazal-oral terapijska shema uključuje dodavanje jednoga bazalnog inzulina najčešće navečer pred spavanje (obično dugodjelujući inzulinski analog) uz postojeću peroralnu terapiju. Obično se to radi kod bolesnika koji imaju povišene vrijednosti glikemije natašte i to je najčešća strategija kojom se započinje inzulinska terapija kod bolesnika sa šećernom bolesti tip 2.
Bazal-plus shema uključuje dodavanje jedne (eventualno dvije) doze brzodjelujućega inzulina prije glavnoga obroka. Obično se radi o bolesnicima koji su na oral-bazal shemi, a imaju postprandijalnu hiperglikemiju i primjenjuje se uglavnom prije glavnoga obroka.
Bazal-bolus shema uključuje primjenu jedne doze dugodjelujućega inzulina, najčešće prije spavanja, čime se osigurava bazalna potreba za inzulinom tijekom 24 sata uz primjenu brzodjelujućih inzulina prije obroka (doručak, ručak i večera). Ovo je klasična shema za primjenu inzulina kod bolesnika sa šećernom bolesti tip 1 te se koristi kao intenzivirana inzulinska terapija za bolesnike sa šećernom bolesti tip 2. Ovaj način primjene inzulina najbliži je fiziološkom obrascu lučenja inzulina, zahtijeva dobru suradljivost bolesnika i veći broj aplikacija inzulina te redovitu kontrolu glikemije.
Liječenje predmiješanim inzulinima označava primjenu predmiješanih inzulina najčešće kao dvije doze (prije doručka i prije večere), rijetko kao tri doze. Predmiješani inzulini sadrže i dugodjelujuću i brzodjelujuću komponentu koje pokrivaju bazalne i postprandijalne potrebe za inzulinom. Ovaj način liječenja obično je rezerviran za starije bolesnike i slabije suradljive bolesnike (zahtijeva manji broj aplikacija inzulina i rjeđe mjerenje glukoze u plazmi).
TERAPIJSKI PRISTUP BOLESNIKU SA ŠEĆERNOM BOLESTI tip 1
U trenutku postavljanja dijagnoze šećerne bolesti tip 1 inzulinopenija je toliko izražena da je inzulinska terapija neophodna u daljnjemu liječenju. Današnje spoznaje sugeriraju primjenu intenzivirane inzulinske terapije po bazal-bolus shemi koja podrazumijeva višekratnu dnevnu primjenu inzulina te redovitu samokontrolu glukoze u plazmi. Druga je opcija primjene inzulinske terapije kod ovih bolesnika primjena inzulina putem inzulinskih pumpi.
Bazal-bolus shema podrazumijeva primjenu dugodjelujućih inzulinskih analoga za bazalne inzulinske potrebe te brzodjelujućih inzulinskih analoga koji pokrivaju prandijalne inzulinske potrebe. Od brozodjelujućih inzulinskih analoga primjenjuju se inzulin lispro, inzulin aspartat ili inzulin glulizin. Regularni ljudski inzulin ne preporuča se za zadovoljavanje prandijalnih inzulinskih potreba. Brzodjelujući inzulinski analozi primjenjuju se tri puta na dan, prije doručka, ručka i večere. Od dugodjelujućih inzulinskih analoga na raspolaganju su inzulin detemir, inzulin glargin U100, inzulin glargin U300 i inzulin degludek. Oni se primjenjuju kako bi zadovoljili bazalne potrebe za inzulinom tijekom 24 sata. Da bi se to ostvarilo, inzulin glargin U300 i inzulin degludek primjenjuju se u jednoj dozi (najčešće pred spavanje), dok inzulin detemir i glargin U100 bolesnici sa šećernom bolesti tip 1 uzimaju u jednoj ili dvije dnevne doze, budući da se pokazalo da njihov učinak traje oko 17 sati. Ta se tri inzulinska analoga ne mogu miješati s ostalim inzulinima i moraju se davati odvojeno od brzodjelujućih inzulinskih analoga. Alternativno se za zadovoljavanje bazalnih inzulinskih potreba može primjenjivati NPH inzulin u manjim dozama prije svakoga obroka (3 - 4 jed), a za razliku od dugodjelujućih inzulinskih analoga može se miješati s brzodjelujućim inzulinskim analozima. Primjer intenzivirane inzulinske terapije prema bazal-bolus shemi za bolesnika sa šećernom bolesti tip 1 prikazana je u Tablici 8.6.
|
Tablica 8.6. Primjeri intenzivirane inzulinske terapije kod bolesnika sa šećernom bolesti tip 1
|
|
Shema
|
Inzulin
|
Prije doručka
|
Prije ručka
|
Prije večere
|
Prije spavanja
|
Izračun se odnosi na prosječnu osobu tjelesne težine od oko 70 kg, uzunos ugljikohidrata raspoređen tako da uzima oko 75 g za doručak, 60 g za ručak i 90 g za večeru. Doza brzodjelujućega inzulina može se povisiti za 1 do 2 ij ako će osoba unijeti više ugljikohidrata u obroku (15 - 30 g) ili je GUP izmjeren prije obroka viši od 9 mmol/L.
|
|
1
|
Brzodjelujući analog
|
5 ij
|
4 ij
|
6 ij
|
--
|
|
Inzulin detemir
|
6 - 7 ij
|
--
|
--
|
8 - 9 ij
|
|
2
|
Brzodjelujući analog
|
5 ij
|
4 ij
|
6 ij
|
--
|
|
Inzulin glargin U100
|
--
|
--
|
--
|
15 - 16 ij
|
Primjena inzulina putem inzulinskih pumpi (CSII, od engl. continuous subcutaneous insulin infusion) predstavlja najfleksibilniji pristup liječenju inzulinom, a podrazumijeva kontinuirano otpuštanje brzodjelujućega inzulina iz uređaja preko katetera s iglom na kraju za postavljanje u supkutano masno tkivo. Uređaj je malen, jednostavan za upotrebu te prijenosan. Kontinuirano otpuštanje malih doza osigurava zadovoljavanje bazalnih inzulinskih potreba, a zadovoljavanje prandijalnih inzulinskih potreba osigurano je dodatnim otpuštanjem potrebne doze inzulina, što programira sama oboljela osoba. Isporuka bazalne doze unaprijed je zadana i osigurava je inzulinska pumpa, a količinu inzulina prije obroka izračunava sam bolesnik. Takav sustav omogućava jednostavniju i bolju kontrolu glikemije (izbjegava se nepredvidivost apsorpcije dugodjelujućega inzulinskog analoga jer se primjenjuju samo brzodjelujući inzulinski analozi čija je apsorpcija predvidljivija) te veću fleksibilnost u svakodnevnome životu (odgoda obroka, raznolikost u veličini i sastavu obroka i sl.). Pri takvoj se shemi primjene inzulina radi boljega praćenja kretanja glikemije mogu koristiti metode kontinuiranoga mjerenja glukoze, bilo minimalno invazivne (supkutane enzimatske elektrode, mikrodijaliza, reverzna ionoforeza), bilo neinvazivne (infracrvena spektroskopija, dielektrična spektroskopija). Glikemija se dakle osigurava isporukom unaprijed određene bazalne doze, bolus doze koju bolesnik sam izračunava i primjenjuje prije obroka te korektivne doze koja se može isporučiti nakon obroka ako glikemija nakon jela nije zadovoljavajuća. Bolesnike je potrebno podučiti o izračunu doze inzulina koju mora primijeniti prije obroka, a ona ovisi o ugljikohidratnim jedinicama.
TERAPIJSKI PRISTUP BOLESNIKU SA ŠEĆERNOM BOLESTI tip 2
Svim bolesnicima sa šećernom bolesti tip 2 koji su pretili treba ukazati na potrebu za smanjenjem tjelesne mase, a to se postiže dijetoterapijom te povećanom tjelesnom aktivnošću. Smanjenjem tjelesne mase smanjuje se inzulinska rezistencija čime se postiže i bolja regulacija glikemije. U farmakološkom liječenju šećerne bolesti tip 2 koriste se oralni i injektabilni antihiperglikemici.
Primjena oralnih antihiperglikemika. Metformin je lijek prvoga izbora ako nije kontraindiciran i ako ga se dobro podnosi. Današnje preporuke sugeriraju da se odmah po postavljanju dijagnoze uvede metformin u terapiju bez čekanja učinka nefarmakoloških mjera. Ako se metformin u klasičnoj dozi teže podnosi (prvenstveno uslijed gastrointestinalnih nuspojava), preporuča se pokušati s primjenom metformina u formulaciji s produljenim oslobađanjem. Ako je metformin kontraindiciran ili se ne podnosi, odabire se bilo koji drugi oralni antihiperglikemik uz poštivanje indvidualnih i drugih karakteristika. Najvažnije karakteristike koje treba uzeti u obzir pri odabiru pojedinih lijekova prikazane su u Tablici 8.7. Ako se monoterapijom ne postigne zadovoljavajuća regulacija glikemije, uz metformin se može dodati još jedan oralni antihiperglikemik. Ako se ni na taj način ne postigne zadovoljavajuća glikemija u razdoblju od tri mjeseca, na raspolaganju su tri mogućnosti: 1) zamjena drugoga oralnog antihiperglikemika drugim - s različitim mehanizmom djelovanja, 2) dodavanje trećega oralnog antihiperglikemika s drugačijim mehanizmom djelovanja ili 3) uvođenje inzulina. Treba imati na umu da pri kombiniranoj terapiji s više hipoglikemika suradljivost bolesnika opada, pa je i regulacija glikemije lošija. Kod pretilih je osoba potrebno izbjegavati lijekove koji dovode do porasta tjelesne mase i birati one sa suprotnim učinkom (metformin, agonisti GLP-1 receptora, SGLT-2 inhibitori). Također, kod osoba kod kojih se u trenu postavljanja dijagnoze bilježe visoke vrijednosti glikiranoga hemoglobina (≥ 9 %) preporuča se odmah početi s dvojnom terapijom oralnim antihiperglikemicima.
|
Tablica 8.7. Karakteristike antihiperglikemika
|
|
Lijek
|
Metformin
|
Sulfonilureja
|
Tiazolidini
|
DPP-4 inhibitori
|
SGLT-2 inhibitori
|
GLP-1 agonisti
|
|
Učinak na glikemiju
|
Visok
|
Visok
|
Visok
|
Srednji
|
Srednji
|
Visok
|
|
Rizik od hipoglikemije
|
Nizak
|
Umjeren
|
Nizak
|
Nizak
|
Nizak
|
Nizak
|
|
Učinak na tjelesnu masu
|
Neutralan/ smanjenje
|
Povećanje
|
Povećanje
|
Smanjenje
|
Smanjenje
|
Smanjenje
|
|
Nuspojave
|
Probavne
Laktacidoza
|
Hipoglikemija
|
Retencija tekućine,
|
Rijetke
Pankreatitis
|
Uroinfekcija
Dehidracija
|
Povraćanje, pankreatitis
|
Liječenje inzulinom. Osnovne indikacije za uvođenje inzulina u liječenje šećerne bolesti tip 2 su: 1) nezadovoljavajuća regulacija glikemije unatoč primjeni oralnih antihiperglikemika i agonista GLP-1 receptora i 2) liječenje inzulinom opravdano je i kao inicijalna terapija uz oralne antihiperglikemike ako su vrijednosti glikiranog hemoglobina visoke (≥ 10 %) pri postavljanju dijagnoze. Pri uvođenju inzulina u terapiju potrebno se držati nekoliko preporuka. Liječenje inzulinom treba započeti niskim dozama (0,1-0,2 jed kg/dan).
Početak inzulinske terapije kod bolesnika sa šećernom bolesti tip 2 preporuča se po bazal-oral shemi. Uvodi se jedna doza bazalnoga inzulina pred spavanje (dugodjelujući inzulinski analozi) uz postojeću terapiju oralnim antihiperglikemicima (najčešće metforminom). Najčešće se primjenjuju inzulin detemir, inzulin glargin U100, glargin U300 i inzulin degludek. Preporučena doza je 0,1 - 0,2 jed/kg. Doza se dalje prilagođava prema vrijednostima glikemije natašte. U slučaju hiperglikemije natašte, doza inzulina se povećava za 2 - 4 jed (ili 10 - 15 % postojeće doze) s razmakom od nekoliko dana (dozu je moguće povisiti jednom ili dva puta u tjedan dana). Ako se razvijaju hipoglikemije, potrebno je smanjiti dozu za 4 jed/kg (ili 10 - 20 % postojeće doze).
Ako se bazal-oral shemom ne postižu željene vrijednosti glikemije, potrebno je intenzivirati inzulinsku terapiju, tj. na raspolaganju su nam tri opcije: bazal-plus shema, liječenje predmiješanim inzulinima dodavanje agonista GLP-1 receptora. Ako se ni time ne postigne željena glikemija, indicirano je intenzivirati inzulinsku terapiju po bazal-bolus shemi.
Bazal-plus shema podrazumijeva dodatak jednoga brzodjelujućeg inzulina prije najvećega obroka uz postojeću oral-bazal shemu. U tu se svrhu upotrebljava regularni (humani) inzulin ili, češće, brzodjelujući inzulinski analozi (inzulin lispro, inzulin aspartat, inzulin glulizin). Taj se način obično preferira kod bolesnika koji imaju uredne vrijednosti glikemije natašte, a unatoč tome ne postižu ciljne vrijednosti glikiranoga hemoglobina. Počinje se s dozom od 4 jed ili 0,1 jed/kg ili 10 % od doze bazalnoga inzulina. Ako se ne postiže bolja glikemija postprandijalno, može se povisiti dozu za 1 - 2 jed (ili 10 - 15 % postojeće doze). U slučaju postprandijalnih hipoglikemija potrebno je dozu sniziti za 1 - 2 jed (ili 10 - 20 % postojeće doze).
Liječenje predmiješanim inzulinima najčešći je oblik intenziviranja inzulinske terapije kod starijih osoba jer zahtijeva manji broj primjena i ustaljen ritam obroka. Premiješani inzulini primjenjuju se u dvije (ili tri) doze, najčešće prije doručka i prije večere. Izbor omjera 25/75, 30/70 ili 50/50 ovisi o vrijednostima glikemije natašte i postprandijalno. Ako je GUP viši natašte, preporuča se primjena pripravka s većim udjelom dugodjelujućega inzulina, a ako je GUP viši postprandijalno, preporuča se primjena pripravka s većim udjelom brzodjelujućega inzulina. Početna doza kod uvođenja prilagođava se postojećoj dozi bazalnoga inzulina, tj. doza bazalnoga izulina se podijeli tako da se dvije trećine te doze primjenjuje ujutro prije doručka, a trećina prije večere. Ako se ne postižu željene vrijednosti glikemije natašte i postprandijalno, doza se povisuje za 1 - 2 jed (ili 10 - 15 % postojeće doze), a povećanje se vrši jednom do dva puta u jednom tjednu. Ako se pojavljuju hipoglikemije, smanjuje se doza za 2 - 4 jed (ili 10 - 20 % postojeće doze).
Dodavanje agonista GLP-1 receptora na bazal-oral shemu novija je metoda intenziviranja inzulinske terapije. Njezinu su učinkovitost pokazale brojne novije studije. Posebno se dobrom pokazuje kod pretilih bolesnika.
Bazal-bolus shema rezervirana je za bolesnike kod kojih se željena regulacija glikemije ne može postići prethodno navedenim shemama. Također, danas se preporučuje i preferira bazal-bolus shema u odnosu na liječenje predmiješanim inzulinima, ali s obzirom na to da je nešto složenija i zahtijeva veću suradljivost bolesnika (veći broj primjena inzulina), nije najbolje rješenje za starije bolesnike. Princip je isti kao i kod bolesnika sa šećernom bolesti tip 1: primjena bazalnoga inzulina (dugodjelujući inzulinski analog) prije spavanja uz primjenu brzodjelujućih inzulina prije glavnih obroka (doručak, ručak, večera). Ukupno potrebna dnevna doza dijeli se u nekoliko doza: 50 % doze daje se kao dugodjelujući inzulin, a ostatak se daje podijeljen u tri doze prije obroka. Doza dugodjelujućega inzulina prilagođava se u odnosu na vrijednosti GUP-a natašte (povećanje ili smanjenje za 2 - 4 jed, do dva puta u tjedan dana). Doza brzodjelujućega inzulina prilagođava se u odnosu na vrijednost glikemije prije sljedećega obroka. Za razliku od šećerne bolesti tip 1, kod koje se doza prandijalnih inzulina računa prema količini ugljikohidrata u obroku, ovdje to uglavnom ne vrijedi, s obzirom na to da ovi bolesnici imaju razvijenu inzulinsku rezistenciju.
Bez obzira na sve navedeno, liječenje šećerne bolesti tip 2 potrebno je prilagoditi individualno, uzimajući u obzir suradljivost bolesnika, kliničke i druge parametre. Shema uvođenja inzulinske terapije bolesnicima sa šećernom bolesti tip 2 prikazana je na Slici 9.
Praćenje bolesnika. Oboljele od šećerne bolesti potrebno je redovno kontrolirati ovisno o načinu liječenja i zadovoljavajućoj regulaciji glikemije. Osim toga, u smislu što ranije dijagnostike i liječenja komplikacija, potreban je probir na kronične komplikacije šećerne bolesti. Preporučene smjernice o praćenju bolesnika sa šećernom bolesti uključuju: redovno samoodređivanje glikemije; određivanje HbA1c (2 - 4 puta godišnje); edukacija bolesnika o liječenju šećerne bolesti; edukacija o prehrani; kompletni oftalmološki pregled (jednom godišnje); pregled stopala (jednom godišnje); određivanje albuminurije (jednom godišnje); mjerenje arterijskoga tlaka (tromjesečno); lipidnoga profila (godišnje) te cijepljenje protiv gripe i pneumokoka.
AKUTNE KOMPLIKACIJE ŠEĆERNE BOLESTI
Među akutne komplikacije šećerne bolesti ubrajamo dijabetičku ketoacidozu, hiperglikemijsko hiperosmolarno stanje te laktacidozu.
DIJABETIČKA KETOACIDOZA
Definicija. Dijabetička ketoacidoza (DKA) predstavlja teški metabolički poremećaj koji nastaje uslijed relativnoga ili apsolutnog manjka inzulina. Svojstven je bolesnicima sa šećernom bolesti tip 1, ali se može javiti i kod bolesnika sa šećernom bolesti tip 2. Osnovna obilježja dijabetičke ketoacidoze su hiperglikemija, hiperketonemija te metabolička acidoza. Radi se o ozbiljnome poremećaju koji zahtijeva što hitnije liječenje. Unatoč poduzetim mjerama liječenja, prosječna smrtnost od dijabetičke ketoacidoze u razvijenim zemljama iznosi 5 –10 %, osobito u starijoj životnoj dobi.
Etiologija. Dijabetička ketoacidoza može biti inicijalno zbivanje i prva manifestacija šećerne bolesti tip 1, ali se može javiti i kod bolesnika s već dijagnosticiranom šećernom bolesti tip 2. U obje se skupine bolesnika javlja pri količini inzulina ispod kritične razine, a to je uglavnom potaknuto nekim zbivanjem u organizmu koje zahtijeva veće količine inzulina što predstavlja točku u kojoj nastaje metabolička dekompenzacija i razvoj dijabetičke ketoacidoze. Najčešći precipitirajući događaji za razvoj dijabetičke ketoacidoze su: 1) neodgovarajuća primjena inzulina; 2) infekcija (pneumonija, genitourinarne infekcije, gastroenteritis, sepsa); 3) kardiovaskularni incident (cerebrovaskularni inzult, infarkt miokarda, mezenterijalna ishemija); 4) lijekovi ili droge (primjerice kokain); 5) trudnoća (Tablica 8.8.).
|
Tablica 8.8. Precipitirajući čimbenici razvoja dijabetičke ketoacidoze
|
|
Česti čimbenici
|
Manje česti čimbenici
|
|
Infekcije (pneumonija, urosepsa, gastroenteritis)
Neodgovarajuća primjena inzulinske terapije
Ishemija ili infarkt miokarda
|
Akutna cerebovaskularna oboljenja
Akutna plućna embolija
Pojačan unos alkohola
Lijekovi (kortikosteroidi, tiazidi, simpatomimetici)
|
Patogeneza. Glavne patofiziološke značajke dijabetičke ketoacidoze su hiperglikemija, hiperketonemija, metabolička acidoza te elektrolitni disbalans. Sve su te promjene potaknute relativnim ili apsolutnim deficitom inzulina uz kompenzatorno povišenje kontraregulatornih hormona (glukagon, katekolamini, kortizol, hormon rasta). Hiperglikemija kao posljedica deficita inzulina dovodi do izražene osmotske diureze kojom se gube voda i elektroliti, posebice natrij i kalij. Gubitak tekućine dovodi do dehidracije.
Manjak inzulina i porast kontraregulatornih hormona dovode do pojačane lipolize pri čemu nastaju slobodne masne kiseline koje odlaze u jetru. Normalno se slobodne masne kiseline u jetri pretvaraju u trigliceride ili lipoproteine vrlo male gustoće (VLDL). Uslijed hiperglukagonemije ovaj proces kreće u smjeru povećane proizvodnje ketonskih tijela zbog aktivacije enzima karnitinske palmitoiltransferaze 1. Taj enzim ima ključnu ulogu u transportu slobodnih masnih kiselina u mitohondrije hepatocita gdje se vrši β-oksidacija i pretvorba u ketonska tijela. U procesu β-oksidacije nastaje acetil-CoA, a kako nastaje više acetil-CoA nego ga se potroši u Krebsovu ciklusu, po dvije molekule acetil-CoA se kondenziraju i nastaje acetooctena kiselina, a iz nje β-hidroksimaslačna kiselina i aceton, ujedno i najznačajnija ketonska tijela. Ketonska tijela ponašaju se kao kiseline, a otklon od fiziološkoga pH krvi sprečavaju bikarbonati koji ih neutraliziraju. Međutim, kako se bikarbonati troše, tako dolazi i do slabljenja toga kompenzatornog mehanizma te se postupno razvija acidoza. Kasnije pogoršanju metaboličke acidoze pridonosi i povećano stvaranje laktata.
Jedan od bitnih događaja u patogenezi dijabetičke ketoacidoze je i nastanak hipokalijemije. Ona je posljedica pojačane diureze, ali i razvoja sekundarnoga hiperaldosteronizma zbog smanjene perfuzije bubrega, uslijed dehidracije. Ipak, koncentracija kalija u ekstracelularnom prostoru rijetko je smanjena, što je posljedica metaboličke acidoze. To znači da kod bolesnika sa slikom dijabetičke ketoacidoze u laboratorijskim nalazima krvi rijetko nalazimo hipokalijemiju. Navedeno je posljedica toga što prilikom acidoze dolazi do ulaska vodikovih iona u stanice uz istodobni izlazak kalija iz stanice, tako da nalazimo smanjenje ukupne količine kalija u organizmu, dok je razina ekstracelularnoga kalija uredna. Ubrzo nakon početka liječenja uz primjenu volumena, koji dovodi do hemodilucije, i nakon primjene inzulina koji dovodi do utoka kalija u stanice, hipokalijemija postaje jasno vidljiva i u ekstracelularnom prostoru (u laboratorijskim nalazima), stoga se s nadoknadom kalija u dijabetičkoj ketoacidozi mora započeti odmah, unatoč urednoj vrijednosti kalija u laboratorijskim nalazima. Bitno je napomenuti da ne postoji čvrsta korelacija između jačine hiperglikemije i težine bolesti. Nekada se ketoacidoza može razviti i pri umjerenoj hiperglikemiji (15 - 20 mmol/L), a nekada se ni uz visoku hiperglikemiju ne razvije ketoacidoza. Također, opisani su i slučajevi tzv. euglikemijske dijabetičke ketoacidoze (opisano kasnije).
Klinička slika. Simptomi i znakovi dijabetičke ketoacidoze razvijaju se postupno, najčešće tijekom 24 sata. Kod bolesnika se pojavljuju kliničke manifestacije bolesti i stanja koja precipitiraju razvoj ketoacidoze, a najčešće se radi o infekcijama. Najraniji simptomi su bol u trbuhu, mučnina i povraćanje. Ti simptomi mogu biti toliko izraženi da nalikuju na kliničku sliku akutnoga abdomena. Bolesnici imaju poliuriju i polidipsiju. Kako se bolest razvija, tako dolazi i do stupnjevitoga poremećaja svijesti (konfuzija, letargija, somnolencija, stupor, koma). Zbog razvoja acidoze javlja se duboko i šumno disanje (Kussmaulovo disanje) kao kompenzacijski mehanizam. Karakterističan zadah bolesnika podsjeća na aceton (trule jabuke). U fizikalnom nalazu vidljivi su hipotenzija, tahikardija, filiformni puls te znakovi dehidratacije (suh i obložen jezik, smanjen turgor kože).
Dijagnoza. Ključna točka postavljanja dijagnoze je postavljanje sumnje na dijabetičku ketoacidozu. To je osobito bitno kada se bolesnici javljaju s dominantnim simptomima bolova u trbuhu, mučnine i povraćanja. Nalaz hiperglikemije, glikozurije i ketonurije zahtijeva određivanje acidobaznoga statusa bolesnika. Sama potvrda dijagnoze dijabetičke ketoacidoze dobiva se iz laboratorijskih nalaza krvi, urina i određivanja plinskih analiza i acidobaznoga statusa. Klasični laboratorijski nalazi uključuju hiperglikemiju, glikozuriju, ketonuriju, sniženje pH arterijske krvi (6,8 - 7,3) uz manjak bikarbonata (< 15 mmol/L). U acidobaznom statusu povećan je anionski zjap. Obvezno je određivanje i elektrolita, posebice natrija i kalija. Najčešće se registrira normokalemija, iako ona ne odražava stvarno stanje kalija u organizmu. Potrebno je provesti i drugu dijagnostiku u smislu otkrivanja precipitirajućega stanja (kompletna krvna slika, diferencijalna krvna slika, C-reaktivni protein, jetreni enzimi, amilaze u serumu i urinu, urea i kreatinin, elektrokardiogram, rendgenska snimka srca i pluća, urinokultura). Česti nalazi u ketoacidozi su i prerenalna azotemija te hiperamilazemija koja nije pankreatičnoga porijekla, na što treba misliti da se ne bi proglasio pankreatitis, osobito kod bolesnika kod kojih je bol u trbuhu jako izražen. Tim je bolesnicima potrebno odrediti i markere srčanoga oštećenja (troponin) zbog visoke učestalosti ishemije miokarda kao provocirajućega čimbenika u razvoju dijabetičke ketoacidoze. Kod tih bolesnika nalazimo i hipertrigliceridemiju kao posljedicu poremećenoga metabolizma masti u nedostatku inzulina. Težinu ketoacidoze najviše procjenjujemo na temelju težine acidoze, a ne na temelju hiperglikemije. Osim toga, superponirana alkaloza koja se može razvijati uslijed jakoga povraćanja može maskirati pravu težinu ketoacidoze. Dijagnostički kriteriji za DKA i HHS prikazani su u Tablici 8.9.
|
Tablica 8.9. Dijagnostički kriteriji za DKA i HHS
|
|
Klinički kriterij
|
Blaga DKA
|
Umjerena DKA
|
Teška DKA
|
HHS
|
|
Glukoza u plazmi
|
> 13,5 mmol/L
|
> 13,5 mmol/L
|
> 13,5 mmol/L
|
> 30 mmol/L
|
|
Osmolalnost seruma
|
Varijabilna
|
Varijabilna
|
Varijabilna
|
> 320 mOsm/kg
|
|
Ketonurija
|
Pozitivna
|
Pozitivna
|
Pozitivna
|
Negativna
|
|
pH arterijske krvi
|
7,25 - 7,35
|
7,00 - 7,24
|
|
> 7,30
|
|
Bikarbonati
|
15 - 18
|
10 - 15
|
< 10
|
> 18
|
|
Anionski zjap
|
> 10
|
> 12
|
> 12
|
< 12
|
|
Mentalni status
|
Budno stanje
|
Somnolencija
|
Sopor, koma
|
Sopor, koma
|
Diferencijalna dijagnoza. Hiperglikemijsko hiperosmolarno stanje nije praćeno ketozom jer u ovom slučaju postoji određena količina inzulina, dostatna za izbjegavanje lipolize, ali ne i za transport glukoze. Alkoholna ketoacidoza nije karakterizirana hiperglikemijom većom od 13 mmol/L. Kod bolesnika koji su na terapiji metforminom u obzir dolazi i laktacidoza. Intoksikacija metanolom, izopropilnim alkoholom i paraledihidom može imati slične simptome i laboratorijske nalaze, ali kod njih ne nalazimo značajniju hiperglikemiju.
Liječenje. Liječenje dijabetičke ketoacidoze treba započeti što ranije kako bi se spriječilo daljnje pogoršanje metaboličkoga poremećaja. Liječenje se provodi u jedinicama intenzivne internističke skrbi. Glavni ciljevi su zadovoljavajuća nadoknada volumena, primjena inzulina te ispravljanje hipokalijemije. Ti postupci obično dovode do rješavanja metaboličke acidoze, a samo u teškim slučajevima indicira se i primjena bikarbonata kako bi se ispravio pH arterijske krvi.
Nadoknada volumena ključna je u liječenju dijabetičke ketoacidoze s obzirom na velike gubitke volumena (deplecija volumena iznosi od 4 do 8 L). Nadoknada se provodi intravenskim primjenama infuzijskih otopina. Preporuča se započeti primjenom fiziološke otopine (0,9 %-tni NaCl) u količini 1 L/h kroz prvih sat do dva sata, a zatim nastaviti brzinom 250 - 500 ml/h. Fiziološku otopinu koristimo sve dok je koncentracija natrija u serumu ispod 150 mEq/L. Kada je koncentracija veća od 150 mEq/L, primjenjuje se 0,45 %-tni NaCl. Tijekom primjene infuzije potrebno je stalno nadgledanje volumnoga statusa, osobito kod starijih osoba, srčanih bolesnika te bolesnika s kroničnom bubrežnom insuficijencijom (opasnost od volumnoga preopterećenja). Pri koncentraciji glukoze u plazmi od 13 do 15 mmol/L potrebno je fiziološku otopinu zamijeniti otopinom 5 %-tne glukoze kako bi se spriječilo nastanak hipoglikemije. Neki autori zagovaraju primjenu otopine 0,45 %-tnoga NaCl-a odmah nakon primjene 1 - 2 L 0,9 %-tnoga NaCl-a, osobito ako se moraju infundirati veće količine volumena s ciljem izbjegavanja naknadne hiperkloremije.
Primjena inzulina u dijabetičkoj ketoacidozi reducira hiperglikemiju (povećana periferna utilizacija glukoze, inhibiranje glukoneogeneze) te pomaže u korekciji acidoze (smanjuje lipolizu, a time i dotok slobodnih masnih kiselina u jetru te ketogenezu). Preporuča se intravenska primjena kratkodjelujućih inzulina kao što su regularni inzulin ili inzulin aspartat, i to putem perfuzora. Danas postoje pripremljene sheme za primjenu inzulina kod bolesnika s dijabetičkom ketoacidozom, a jedna od njih uključuje miješanje 50 jedinica inzulina i 50 mililitara fiziološke otopine tako da jedan mililitar sadrži jednu jedinicu inzulina, a sam perfuzor osigurava preciznu aplikaciju određene (zadane) doze inzulina tijekom jednoga sata. Primjena inzulina u infuzijskoj otopini manje je pouzdan način da se primijeni potrebna doza inzulina u određenome vremenskom intervalu, osim toga, odvojena primjena inzulina preko perfuzora omogućava neovisnu promjenu doza izulina ili količine infudirane tekućine. Postoje dvije strategije pri doziranju inzulina na početku liječenja dijabetičke ketoacidoze: 1) početno se primjenjuje doza zasićenja od 0,1 jed/kg u obliku intravenskoga bolusa, a zatim se nastavlja s kontinuiranom primjenom inzulina u dozi od 0,1 jed/kg/h ili 2) inzulin se ordinira u dozi od 0,14 jed/kg/h bez inicijalnoga intravenskog bolusa. To su dostatne vrijednosti inzulina, dovoljne za postizanje željenoga učinka kod većine bolesnika. Nastavak doziranja inzulina ovisi o padu vrijednosti glukoze u prvih sat vremena. Treba izbjegavati naglo snižavanje glukoze u plazmi zbog mogućih komplikacija, kao što je moždani edem, a preporuka je da se glikemija snižava za 3 - 4 mmol/L na sat ili snižavanje za oko 10 % u odnosu na početnu vrijednost. Ako je brzina snižavanja glukoze veća od preporučenoga, potrebno je smanjiti dozu primjenjivanoga inzulina, a ako to preporučeno sniženje glukoze izostane u prvih sat vremena, preporuča se primjena intravenskoga bolusa od 0,1 jed/kg te nastavak kontinuirane primjene. Kada vrijednost glukoze u plazmi dosegne vrijednost između 13 i 15 mmol/L, smanjuje se doza inzulina na 0,05 jed/kg/h. Bolesnici koji inače u terapiji imaju dugodjelujuće inzulinske analoge (inzulin glargin U100, inzulin detemir, inzulin glargin U300 ili inzulin degludek) trebaju nastaviti s njihovom primjenom kao i ranije. U rijetkim slučajevima imunološki posredovane inzulinske rezistencije liječenje se provodi udvostručavanjem doze inzulina svaka 2 do 4 sata dok se ne postigne željeni učinak. Kao alternativa intravenskoj primjeni inzulina, preporuča se intramuskularna primjena (inicijalna doza 20 - 20 jedinica koju slijedi doza od pet jedinica svakih sat vremena) ili supkutana primjena (inicijalna doza od 0,3 jed/kg te nastavak 0,1 jed/kg svakih sat vremena).
Nadoknada kalija također je sastavni dio liječenja, s obzirom na to da njegov ukupni deficit može iznositi i do 200 mEq uslijed poliurije i povraćanja. Kako je već opisano, laboratorijske vrijednosti kalija unatoč tome ostaju normalne ili čak povišene, zbog pomaka kalija iz intracelularnoga prostora uslijed acidoze. Pad u laboratorijskim vrijednostima nastaje nakon početka liječenja uslijed dilucije primjenom infuzijskih otopina te pomakom kalija u stanice uslijed korekcije acidoze. Svim bolesnicima čiji je kalij ispod 5,5 mmol/L i koji nemaju znakova bubrežnoga zatajenja kalij se nadoknađuje u sljedećim dozama: ako je kalij ispod 3,5 mmol/L, dodaje se 40 ml 7,4 %-tnoga KCl-a u svaku litru infuzije; ako je kalij između 3,5 i 4,4 mmol/L, dodaje se 30 ml 7,4 %-tnoga KCl-a u svaku litru infuzije; a ako je kalij između 4,5 i 5,5 mmol/L, dodaje se 20 ml 7,4 %-tnoga KCl-a u svaku litru infuzije. Ako je kalij iznad 5,5 mmol/L, ne nadoknađuje se, nego se samo prati njegova vrijednost i postupa se prema novoj vrijednosti. Kalij se prati svakih 4 do 6 sati. Kod primjene kalija bitno je pratiti i diurezu i uočiti potencijalni razvoj bubrežne insfucijencije jer se tada obustavlja primjena kalija. Kada se stanje bolesnika popravi te tekućinu počne uzimati peroralno, preporuča se nastaviti uzimanje hrane bogate kalijem, kao što je banana (srednje velika banana sadrži oko 10 mEq kalija) ili sok od rajčice (250 ml sadrži 14 mEq kalija).
Primjena bikarbonata nije opravdana kao rutinski dio liječenja dijabetičke ketoacidoze. Rutinska primjena bikarbonata povezana je s lošijim ishodom bolesti iz nekoliko razloga: 1) brza korekcija acidoze dovodi do brzoga razvoja hipokalijemije zbog unutarstaničnoga pomaka kalija; 2) pogoršava se tkivna hipoksija zbog reducirane disocijacije kisika iz hemoglobina te 3) paradoksalne pojave cerebralne acidoze. Primjena bikarbonata indicirana je kada je pH arterijske krvi ispod 6,9. U tom slučaju primjenjuje se 100 mmol (dvije ampule) bikarbonata u 1000 ml 0,45 %-tnoga NaCl-a uz dodatak 20 ml 7,4 %-tnoga KCl-a tijekom dva sata. Dodatak bikarbonata u otopinu 0,9 %-tnoga NaCl-a stvara hipertoničnu otopinu koja može pogoršati hiperosmolarno stanje pa se to izbjegava. Ova se formulacija primjenjuje se sve dok pH ne poraste iznad 7,0.
Supkutana primjena inzulina. Nakon što se poprave laboratorijski nalazi (glikemija, pH, bikarbonati) te se bolesnik razbudi, može započeti peroralni unos hrane i tekućine te se zaustavlja intravenska primjena volumena, inzulina i kalija. Daljnji postupak s bolesnikom ovisi o tome radi li se o šećernoj bolesti tip 1 ili 2. Kod bolesnika sa šećernom bolesti tip 1 potrebno je prevođenje na supkutanu primjenu inzulina i to po shemi bazal-bolus. Ukupna doza inzulina za te bolesnike uglavnom iznosi 0,5 - 0,8 jed/kg/dan, 50 % te doze daje se u obliku večernje doze dugodjelujućega inzulinskog analoga, a ostatak kao prandijalni brzodjelujući inzulin prije doručka, ručka i večere. Kod tih se bolesnika kroz nekoliko dana smanjuje inzulinska rezistencija, pa im trebaju i manje doze inzulina, stoga se moraju češće kontrolirati te sukladno tome smanjivati dozu inzulina. Bolesnici sa šećernom bolesti tip 2 također nakon poboljšanja postaju kandidati za inzulinsku terapiju, bilo po shemi bazal-bolus, kako je prethodno opisano, bilo uvođenjem terapije predmiješanim inzulinom (ukupna dnevna doza inzulina daje se u dvije doze: dvije trećine doze daje se ujutro pred doručak, a trećina navečer pred večeru). Kod manjega se broja takvih bolesnika nakon rješavanja uzročne bolesti može ponovno prijeći na liječenje oralnim hipoglikemicima bez inzulina. Pri prebacivanju bolesnika s intravenske primjene inzulina na supkutanu, mora se spriječiti nastanak perioda bez pokrivenosti inzulinom. Intravenski unesen inzulin ima poluvijek života od 2,5 minute pa se nakon prestanka primjene vrlo brzo razvija ponovni deficit inzulina, što može uzrokovati relaps ketoacidoze. Iz tih je razloga potrebno supkutano primijeniti inzuline nekoliko sati prije nego se isključi intravenska primjena inzulina. Obično se to provodi tako da bolesnik dobije bazalni i prandijalni inzulin s prvim obrokom te se nakon sat vremena prestane s intravenskom primjenom inzulina.
Ostale napomene u vezi liječenja dijabetičke ketoacidoze uključuju provođenje terapije za osnovnu bolest (najčešće uvođenje antibiotske terapije), primjenu fosfata kod izražene hipofosfatemije (iako studije nisu pokazale veću kliničku korist, a moguć je razvoj hipokalcemije) te ostalih potpornih metoda ako je potrebno. Kod hiperosmolarnosti, tj. kada je osmolalnost seruma iznad 350, ako se računa po formuli 2 x (natrij + glukoza), primjenjuje se tromboprofilaksa uz primjenu niskomolekularnoga heparina (enoksaparin 1 mg/kg supkutano jednom dnevno).
Komplikacije. Najozbiljnija komplikacija dijabetičke ketoacidoze je nastanak moždanoga edema, povezanoga uz nekoliko čimbenika: teška acidoza u trenutku postavljanja dijagnoze, brza korekcija hiperglikemije te ekscesivna nadoknada volumena u prva četiri sata. Kod svih bolesnika kod kojih se u prvim satima terapije pogoršava mentalni status uz pojavu glavobolje, treba misliti na ovo stanje. Dijagnoza se potvrđuje slikovnim metodama (kompjuterizirana tomografija, magnetska rezonancija). Liječenje podrazumijeva intravensku primjenu manitola (1 - 2 g/kg tijekom 15 minuta). Prekomjerna primjena infuzija može dovesti do volumnoga opterećenja i razvoja plućnoga edema, osobito kod srčanih bolesnika. Akutni respiratorni distres sindrom rijetka je komplikacija terapije dijabetičke ketoacidoze.
Prognoza. Pravilno liječenje i praćenje bolesnika smanjilo je smrtnost ovoga stanja na manje od 5 %, no kod starijih bolesnika te bolesnika s predisponirajućim čimbenicima za nastanak dijabetičke ketoacidoze smrtnost je i dalje visoka i iznosi oko 20 %. Nakon oporavka je potrebna edukacija bolesnika o prepoznavanju ranih simptoma ketoacidoze kako bi se spriječilo nastanak nove epizode.
Euglikemijska dijabetička ketoacidoza definira se kao stanje kojega čini metabolička acidoza s povećanim anionskim rascjepom te ketonurija uz gotovo uredne vrijednosti glikemije, odnosno vrijednosti glikemije nižih od uobičajenih za dijabetičku ketoacidozu. S obzirom na takve vrijednosti glikemije, ovo se stanje se može previdjeti u svakodnevnoj kliničkoj praksi. Rijetko se može razviti kod bolesnika koji nemaju zadovoljavajući unos ugljikohidrata u organizam (gladovanje) ili uslijed smanjene proizvodnje glukoze u jetri ili pojačanoga gubitka putem urina (relativni manjak inzulina, porast koncentracije kontraregulatornih homona). Radi se obično o bolesnicima s već ranije dijagnosticiranom šećernom bolesti koji primaju inzulinsku terapiju, u stanjima koja mogu precipitirati nastanak dijabetičke ketoacidoze. Najčešća stanja kod kojih možemo dijagnosticirati euglikemijsku dijabetičku ketoacidozu su smanjen unos ugljikohidrata, trudnoća, pankreatitis, intoksikacija alkoholom, produljen period mučnine i povraćanja, primjena inzulinske pumpe te primjena SGLT2 inhibitora. Kako se to može dogoditi i kod stanja akutne alkoholne intoksikacije, lako se može zamijeniti za alkoholnu ketoacidozu, a moguće ih je razlikovati jedino prema nalazu glikemije: kod euglikemijske dijabetičke ketoacidoze vrijednosti glukoze u plazmi su uredne ili povišene, dok su u alkoholnoj ketoacidozi obično snižene. Liječenje se provodi kao i kod klasične dijabetičke ketoacidoze uz odgovarajuću primjenu inzulina.
HIPERGLIKEMIJSKO HIPEROSMOLARNO STANJE
Definicija. Hiperglikemijsko hiperosmolarno stanje akutna je komplikacija šećerne bolesti uslijed relativnoga manjka inzulina, a očituje se hiperglikemijom, hiperosmolarnošću i dehidracijom, bez značajnije ketoze i metaboličke acidoze. Javlja se pretežno kod starijih osoba sa šećernom bolesti tip 2. Hiperglikemijsko hiperosmolarno stanje susreće se rjeđe od dijabetičke ketoacidoze.
Etiopatogeneza. Kao i dijabetička ketoacidoza, hiperglikemijsko hiperosmolarno stanje potaknuto je nekim precipitirajućim stanjem, kao što su infarkt miokarda, cerebrovaskularni inzult, infekcije (pneumonija, sepsa) ili primjena određenih lijekova (fenitoin, diazoksid, kortikosteroidi). Relativni inzulinski manjak odgovoran je za nastanak hiperglikemije zbog smanjene periferne utilizacije glukoze te povećane produkcije glukoze u jetri (glikogenoliza, glukoneogeneza). Hiperglikemija dovodi do porasta osmolalnosti seruma i uzrokuje osmotsku diurezu što dovodi do deplecije intravaskularnoga volumena i dehidracije, a dehidraciji pogoduje i smanjen unos tekućine koji je čest kod ovih bolesnika. Razlog zbog kojega se kod ovih bolesnika ne razvija hiperketonemija i acidoza nije u potpunosti jasan. Vjerojatno objašnjenje se najvjerojatnije može pronaći u činjenici da kod ovih bolesnika ne nalazimo apsolutni deficit inzulina pa je preostali inzulin dostatan da spriječi lipolizu, ali ne i da održava perifernu utilizaciju. Također, kod ovih bolesnika snižena je koncentracija kontraregulatornih hormona u odnosu na dijabetičku ketoacidozu. Ako ovo stanje potraje duže vrijeme, deplecija intravaskularnoga volumena dovest će do periferne hipoksije s posljedičnim stvaranjem laktata koji mogu provocirati srednje izraženu metaboličku acidozu sa smanjenim anionskim zjapom (engl. anion gap).
Klinička slika. Hiperglikemijsko hiperosmolarno stanje razvija se kroz dulje vrijeme. Obično se radi o starijim bolesnicima s anamnezom poliurije i gubitka na tjelesnoj masi kroz nekoliko tjedana. Razvijaju se umor i malaksalost koji napreduju, pojavljuju se konfuzija i letargija te na kraju stupor i koma. Klasični simptomi dijabetičke ketoacidoze kao što su bol u trbuhu, mučnina, povraćanje i Kussmaulovo disanje, odsutni su. U razvijenom obliku bolesnici su poremećenoga stanja svijesti, oligurični, hipotezivni i tahikardični. U nastavku se, ako se bolest ne liječi, razvija hipovolemični šok.
Dijagnoza. Dijagnoza se postavlja na osnovi anamnestičkih i kliničkih podataka te laboratorijskih vrijednosti. Klasično kod ovih bolesnika nalazimo izraženu hiperglikemiju (GUP > 30 mmol/L), izraženu hiperosmolalnost seruma (> 310 mOsm/kg) uz uredan pH arterijske krvi (> 7,3), uredne bikarbonate (> 15 mEq/L) i normalan anionski zjap (< 14 mEq/L). Natrij u početku može biti snižen (hiperglikemija dovodi do pojačanoga navlačenja tekućine u intravaskularni prostor pa nastaje dilucijska hiponatremija), no s razvojem deplecije volumena i dehidracije raste koncentracija natrija (i više od 140 mEq/L). Ukupna količina kalija u organizmu smanjena je zbog njegova gubitka urinom, a početna laboratorijska vrijednost kalija je normalna ili snižena, za razliku od dijabetičke ketoacidoze (izostanak učinka acidoze na pomak kalija iz stanica). Kod ovih bolesnika razvija se i prerenalna azotemija uz karakteristični porast ureje (iznad 30 mmol/L). Kao i kod dijabetičke ketoacidoze, potrebno je obaviti dijagnostičke pretrage usmjerene na otkrivanje uzročnoga čimbenika koji je provocirao hiperglikemijsko hiperosmolarno stanje (kompletna i diferencijalna krvna slika, biokemijske pretrage krvi, troponin, elektrokardiogram, rendgenske snimke srca i pluća, urinokultura i dr.).
Liječenje. Slično kao i kod dijabetičke ketoacidoze, terapija je usmjerena na nadoknadu volumena, inzulina i kalija. Ostale mjere uključuju tromboprofilaksu niskomolekularnim heparinom, s obzirom na mogućnost nastanka tromboze uslijed hiperosmolarnosti i hemokoncentracije, primjenu antiobiotika kod perzistirajućih infekcija i sl.
Nadoknada volumena ključan je terapijski korak u ovom stanju, s obzirom na to da je dehidracija glavni potencijalno ugrožavajući događaj. Deficit volumena može iznositi i do 10 L tekućine. U hiperglikemijskom hipersomolarnom stanju preporučuje se primjena 0,45 %-tne otopine NaCl-a, da se ne pogoršava hiperosmolarnost s obzirom na to da su tjelesne tekućine već same po sebi hipertonične. U slučaju oligurije i hipotenzije preporuča se primjena 0,9 %-tne otopine NaCl-a. Preprouča se primjena 4 - 6 L volumena u prvih 8 do 10 sati liječenja, a sama brzina volumne nadoknade slična je kao kod dijabetičke ketoacidoze (1000 ml u prvih sat vremena, a potom nastaviti 500 ml na sat) uz praćenje hemodinamskih parametara kako bi se izbjeglo volumno preopterećenje, osobito kod starijih i srčanih bolesnika. Kao i kod dijabetičke ketoacidoze, kada vrijednost GUP-a dosegne 13 - 15 mmol/L potrebno je uključiti 5 %-tnu glukozu. Ovo stanje glikemije oko 15 mmol/L potrebno je održavati neko vrijeme kako bi se izbjegle nagle promjene u osmolalnosti, tj. da bi se preveniralo nastanak moždanoga edema.
Nadoknada inzulina vrši se po principu kao i kod dijabetičke ketoacidoze. Preporuča se primjena brzodjelujućih inzulina preko perfuzora (50 jedinica u 50 ml fiziološke otopine). Inicijalna doza inzulina iznosi 0,1 jed/kg kao intravenski bolus, a potom se nastavlja s 0,1 jed/kg/h u kontinuiranoj intravenskoj primjeni (ili 0,14 jed/kg/h bez inicijalnoga bolusa). Teži se postupnome snižavanju hiperglikemije brzinom 3 do 4 mmol/L. Kada se se glikemija poboljša i kada se vrijednosti kreću oko 15 mmol/L, potrebno je smanjiti dozu intravenskoga inzulina na 0,05 jed/kg/h. Kada bolesnik može jesti, počinje se s prelaskom na supkutanu primjenu.
Nadoknada kalija vrši se kod svih bolesnika, kao i kod dijabetičke ketoacidoze, a doziranje se prilagođava laboratorijskim vrijednostima kalija, kao što je opisano kod dijabetičke acidoze. Kod primjene kalija treba biti oprezan kod bolesnika s renalnom insuficijencijom.
Komplikacije. Teška dehidracija i deplecija intravaskularnoga volumena mogu biti precipitirajući čimbenici razvoja teških komplikacija, kao što su infarkt miokarda, cerebrovaskularni inzult, plućna embolija, mezenterijalna ishemija i diseminirana intravaskularna koagulopatija. Kod nekih se bolesnika bilježi i veća učestalost rabdomiolize za vrijeme liječenja, pa je potrebno pratiti vrijednost kreatin kinaze.
Prognoza. Hiperglikemijsko hiperosmolarno stanje ima lošiju i težu prognozu od dijabetičke ketoacidoze, a dijelom to proizlazi iz činjenice da je ono češće kod starijih osoba s brojnim komorbiditetima. Rano postavljena dijagnoza i započeto liječenje čimbenici su koji povećavaju preživljenje. Nakon stabilizacije bolesnika inzulinska se terapija provodi još nekoliko tjedana te je moguće bolesnike vratiti na oralne antihiperglikemike i dijetoterapiju kao metodu liječenja, ako se popravi endogena inzulinska sekrecija.
LAKTACIDOZA
Definicija. Laktacidoza je karakterizirana nakupljanjem mliječne kiseline u krvi, što dovodi do sniženja pH arterijske krvi i kompenzatornoga sniženja serumskog bikarbonata. Laktacidoza je rijetka kod oboljelih od šećerne bolesti, a može biti sastavni dio ketoacidoze ili se može javiti kod bolesnika koji su na terapiji metforminom (metforminska laktacidoza).
Etiopatogeneza. Normalno su glavni izvor mliječne kiseline eritrociti, skeletni mišići, koža i mozak. Konverzija mliječne kiseline u glukozu i njezina oksidacija u najvećem se dijelu odvija u jetri, ali može i u bubrezima i to predstavlja glavne puteve eliminacije. Laktacidoza može nastati zbog prekomjernoga stvaranja laktata (tkivna hipoksija) ili zbog oslabljene eliminacije (jetrena insuficijencija). Laktacidoza se najčešće susreće kod teško oboljelih osoba (cirkulatorni kolaps – šok, respiratorna insuficijencija, jetrena insuficijencija, septikemija, mezenterijalna ishemija i dr.). Kod bolesnika sa šećernom bolesti laktacidoza je rijetka, ali može biti povezana s primjenom metformina, osobito kod bolesnika koji imaju neki stupanj bubrežnoga zatajenja.
Klinička slika. S obzirom na to da se laktacidoza javlja sekundarno, uz stanja teških oboljenja, sama klinička slika ovisi o uzročnoj bolesti, a uglavnom se radi o teškome stanju (hipotenzija, tahikardija, oligurija, cijanoza, poremećaj svijesti). Glavni znak razvoja laktacidoze je hiperventilacija koja se razvija kao kompenzatorni mehanizam. Kod dijabetičara kod kojih se laktacidoza razvija uslijed primjene metformina nastup simptoma je polaganiji, počinje kao nedefinirano opće loše stanje praćeno umorom i malaksalošću, a potom se postupno razvija prostracija (mučnina, povraćanje, poremećaj svijesti, hipotenzija, oligurija).
Dijagnoza i diferencijalna dijagnoza. Uz anamnestičke i kliničke podatke, laboratorijski su nalazi glavni temelj dijagnoze. Klasični laboratorijski nalazi uključuju pH arterijske krvi ispod 7,3; serumske bikarbonate ispod 15 mEq/L; anionski zjap veći od 15 mEq/L, nedostatak ketonemije i ketonurije te vrijednost laktata veću od 5 mmol/L. Potrebno je provoditi i dodatnu dijagnostiku ovisno o osnovnoj bolesti. Metabolička acidoza uz porast laktata može ići uz dijabetes (hiperketonemija), uremiju (nakupljanje anorganskih kiselina), trovanje lijekovima (salicilati, metanol, etanol) te ingestiju kiselina (acetooctena kiselina i sl.).
Liječenje se provodi liječenjem osnovnoga uzroka bolesti. Glavna metoda liječenja laktacidoze je uspostava zadovoljavajuće tkivne perfuzije (stabilizacija hemodinamskih i respiratornih parametara). Svim bolesnicima koji manifestiraju laktacidozu kojoj se ne može otkriti uzrok daju se antibiotici širokoga spektra zbog sumnje na septičko stanje. Ako postoje drugi metabolički otkloni, potrebno ih je liječiti (hiperglikemija, hipokalijemija i sl.) Primjena bikarbonata rezervirana je za teške oblike, kada pH padne ispod 6,9 mmol/L, a sama primjena je kontroverzna i nije dokazano da poboljšava ishod. Agresivna primjena bikarbonata uključuje primjenu i do 2000 mEq kroz 24 sata. Kod sumnje na metaboličku acidozu uzrokovanu metforminom indicirano je provesti hemodijalizu. Prognoza ovih bolesnika je teška, a ponajprije ovisi o uzročnoj bolesti.
KRONIČNE KOMPLIKACIJE ŠEĆERNE BOLESTI
Kronične komplikacije šećerne bolesti predstavljaju značajan javnozdravstveni problem koji susrećemo u svakodnevnoj praksi. Njihova pojavnost ovisi o trajanju i regulaciji šećerne bolesti, češće se javljaju kod osoba s loše reguliranom šećernom bolesti kroz duže vrijeme. Kronične komplikacije šećerne bolesti u današnje su vrijeme velik problem, glavni su uzrok obolijevanja (morbiditeta) i smrtnosti (mortaliteta) osoba sa šećernom bolesti, a značajno utječu na kvalitetu života, kao i na cijenu liječenja. Velike randomizirane kliničke studije pokazale su da redukcija kronične hiperglikemije značajno odgađa pojavu kroničnih komplikacija, kako pri šećernoj bolesti tip 1, tako i tip 2.
Akademski, kronične komplikacije šećerne bolesti dijelimo u dvije skupine: vaskularne i nevaskularne komplikacije. Vaskularne komplikacije prema zahvaćenosti krvnih žila dijelimo na mikrovaskularne (retinopatija, nefropatija, neuropatija) te makrovaskularne komplikacije (koronarna bolest, periferna bolest arterija). U nevaskularne komplikacije ubrajamo brojna stanja, kao što su promjene na probavnom sustavu, koži i sluznicama, koštanozglobnome sustavu i dr. Mikrovaskularne komplikacije svojstvene su i šećernoj bolesti tip 1 i tip 2, dok se makrovaskularne komplikacije uglavnom povezuju sa šećernom bolesti tip 2.
PATOFIZIOLOGIJA KRONIČNIH KOMPLIKACIJA ŠEĆERNE BOLESTI
Patogeneza kroničnih komplikacija šećerne bolesti kompleksna je i slabo razjašnjena. Patofiziološki gledano, polazna osnova za razvoj komplikacija leži u dugotrajnoj hiperglikemiji, iako nisu još poznati točni mehanizmi kojima ona dovodi do staničnoga oštećenja (stanično oštećenje inducirano glukozom). Postoji više teorija kojima se pokušava objasniti učinak hiperglikemije na staničnu disfunkciju i oštećenje ciljnih stanica: nakupljanje krajnjih produkata glikozilacije, nakupljanje sorbitola, nakupljanje diacilglicerola te aktivacija heksozaminskoga puta.
Nakupljanje krajnjih produkata glikozilacije. Povećana koncentracija glukoze dovodi do formiranja krajnih produkata glikozilacije (AGEs, od engl. advanced glycosylation end products) koji se nakupljaju u mnogim tkivima i organima gdje se vežu za specifični receptor (RAGE). To vezanje aktivira različite intracelularne puteve koji mogu dovesti do raznih učinaka s reperkusijama na staničnu disfunkciju: stimulacija oksidativnih reakcija, pojačano stvaranje proupalnih citokina, pojačano stvaranje faktora rasta i sl.
Nakupljanje sorbitola. Iako je dominantan put metaboliziranja glukoze fosforilacija i glikoliza u njezinom suvišku, dio glukoze metabolizira se u sorbitol uz posredstvo enzima aldoza reduktaze. Povećana intracelularna koncentracija sorbitola mijenja redoks-potencijal stanice, povećava staničnu osmolalnost te generira nastanak slobodnih kisikovih radikala, a sve to vodi prema staničnoj disfunkciji. Ipak, primjena inhibitora aldoza reduktaze u kliničkim ispitivanjima kod bolesnika sa šećernom bolesti nije bila praćena značajnijim kliničkim učinkom glede prevencije i liječenja dijabetičke retinopatije, neuropatije ili nefropatije.
Nakupljanje diacilglicerola. Hiperglikemija također može dovesti do pojačanoga intracelularnog stvaranja i nakupljanja diacilglicerola što vodi prema aktivaciji protein kinaze C. Protein kinaza C, između ostaloga, može dovesti do promjene u transkripciji gena za fibronektin, kolagen tip IV, kontraktilnih proteina te proteina izvanstaničnoga matriksa, posebice u endotelnim stanicama i neuronima. Učinak inhibitora protein kinaze C u kliničkoj praksi još se ispituje.
Aktivacija heksozaminskoga puta. Ova teorija podrazumijeva da hiperglikemija potiče metabolizam glukoze putem heksozaminskoga puta što generira nastanak fruktoza-6-fosfata, supstrata koji ima sposobnost glikozilacije proteina, kao što je endotelna NO sinteza koja ima značajnu ulogu u endotelnoj disfunkciji. Također, aktivacija heksozaminskoga puta može dovesti i do promjene genske ekspresije transformirajućega faktora rasta (TGF-β) ili inhibitora aktivatora plazminogena (PAI-1).
Navedeni poremećaji ponajviše su svojstveni endotelnim stanicama i neuronima, što i objašnjava vrste komplikacija poglavito vezane uz krvne žile (mikroangiopatije, makroangiopatije) te živčane stanice (periferna ili autonomna neuropatija). Sažet prikaz mogućih patofizioloških zbivanja u razvoju kroničnih komplikacija šećerne bolesti prikazan je shematski na Slici 8.2.
Slika 8.2. Patofiziologija kroničnih komplikacija šećerne bolesti
DIJABETIČKA RETINOPATIJA
Definicija. Dijabetička retinopatija kronična je komplikacija šećerne bolesti karakterizirana promjenama na krvnim žilama mrežnice koje se klinički očituju postupnim i progresivnim gubitkom vida. Ovisno o histopatološkom i kliničkom nalazu, razlikujemo dva oblika: neproliferativni i proliferativni oblik. Dijabetička je retinopatija jedan od najčešćih uzroka sljepoće kod odraslih osoba u dobi između 30 i 65 godina u razvijenim zemljama. Ta je komplikacija svojstvena oboljelima i od šećerne bolesti tip 1 i tip 2. Smatra se da oko 20 % bolesnika sa šećernom bolesti tip 2 ima neki oblik dijabetičke retinopatije u trenutku postavljanja dijagnoze.
Etiopatogeneza. Glavni etiološki čimbenici su dužina bolesti, loša kontrola glikemije, hipertenzija, trudnoća, uzimanje oralnih kontraceptiva, pušenje te prekomjeran unos alkohola. Hiperglikemija dovodi do niza biokemijskih promjena u endotelnim stanicama krvnih žila retine što dovodi do endotelne disfunkcije, povećane vaskularne permeabilnosti, poremećene autoregulacije, povećane prozvodnje vazoaktivnih tvari te faktora rasta kao što je faktor rasta vaskularnoga endotela (VEGF).
Neproliferativna dijabetička retinopatija obuhvaća rane promjene u razvoju dijabetičke retinopatije koje nastaju uslijed povećane permeabilnosti kapilarne stijenke, a uključuju nastanak mikroaneurizama, pojavu sitnih točkastih krvarenja te nastanak eksudata (depoziti proteina, lipida i krvnih stanica), što uzrokuje nastanak edema. Kada se takve promjene dominantno odvijaju u području makule, govorimo o makularnome edemu, povezom s oštećenjem vidne oštrine. Prevalencija neproliferativne retinopatije kod bolesnika sa šećernom bolesti tip 2 nakon 15 godina iznosi oko 60 %.
Proliferativna dijabetička retinopatija označava rast novih kapilara (proliferacija) i fibrozu unutar mrežnice te urastanje u staklasto tijelo. To je stanje povezano s hipoksijom kao glavnim stimulirajućim čimbenikom vaskularne proliferacije. Novonastale kapilare su krhke, lako pucaju, i tako nastaju krvarenja, osobito u staklastom tijelu. Neovaskularizacija i fibroza dovode do narušavanja normalnih anatomskih odnosa, što se može manifestirati abrupcijom retine kao najgorim ishodom.
Klinička slika. Neproliferativna dijabetička retinopatija ne dovodi do težih poremećaja vida, osim ako se ne radi o makularnom edemu koji dovodi do smanjenja oštrine vida i iskrivljenja slike. S nastankom i napredovanjem proliferativne retinopatije dolazi do smanjenja vida koje postaje progresivno i može završiti sljepoćom. Oboljeli često opisuju pojavu mrlja ispred zahvaćenoga oka.
Dijagnoza se postavlja na temelju sveobuhvatnoga oftalmološkog pregleda koji uključuje ispitivanje vida (visus), pregled očne pozadine (fundus), procjenu vidne oštrine, mjerenje očnoga tlaka, pregled biomikroskopom, a ponekada se poseže i za sofisticiranijim metodama kao što su optička koherentna tomografija (OCT) ili fluoresceinska angiografija. Bolesnicima sa šećernom bolesti preporuča se jednom godišnje obaviti pregled kod oftalmologa, ako nemaju razvijenu dijabetičku retinopatiju koja zahtijeva češću kontrolu.
Liječenje ovisi o stadiju retinopatije. Osnovni cilj liječenja postizanje je dobre regulacije glikemije kako bi se odgodio nastanak retinopatije ili usporilo njezino napredovanje. Paradoksalno, u prvih 6 do 12 mjeseci od poboljšanja kontrole glikemije (npr. nakon početka liječenja) dolazi do pogoršanja promjena u sklopu retinopatije, ali to je samo privremeno i prolazno. Liječenje fotokoagulacijom najčešća je i najprimjenjivanija metoda liječenja promjena na retini, a obično se u tu svrhu primjenjuje argon ili ksenonski laser. U posljednje se vrijeme sve više primjenjuje anti-VEGF terapija, odnosno intraokularna aplikacija anti-VEGF faktora (bevacizumab) čime se usporava ili zaustavlja neovaskularizacija. Krajnju opciju liječenja predstavlja kirurško liječenje – vitrektomija. Osim dobre regulacije glikemije, pacijentima se savjetuje prestanak pušenja i dobra regulacija arterijskoga tlaka.
Ostale okularne promjene vezane uz šećernu bolest uključuju dijabetičku kataraktu i glaukom. S obzirom na neenzimatsku glikozilaciju proteina leće, staračka mrena (katarakta) dva puta je češća kod bolesnika sa šećernom bolešću u odnosu na nedijabetičku populaciju. Glaukom se javlja kod oko 6 % bolesnika sa šećernom bolesti. Najčešće se povezuje s promjenama uslijed neovaskularizacije i fibroze što vodi prema promjenama u području kuta šarenica i mogućega otežanog dreniranja očne vodice. Dijagnostika i liječenje u nadležnosti su oftalmologa.
DIJABETIČKA NEFROPATIJA
Definicija. Dijabetička nefropatija važan je uzrok morbiditeta i mortaliteta bolesnika sa šećernom bolesti. Smatra se da je ukupno jedna trećina kroničnoga bubrežnog zatajenja uzrokovana dijabetičkom nefropatijom. Bolesnici sa šećernom bolesti tip 1 imaju 30 – 40 % veće šanse za razvoj dijabetičke nefropatije nakon 20 godina oboljenja, dok bolesnici sa šećernom bolesti tip 2 imaju manji rizik, negdje oko 15 do 20 %. Ipak, s obzirom na veći broj bolesnika sa šećernom bolesti tip 2, prevalencija dijabetičke nefropatije veća je kod bolesnika s tipom 2.
Etiopatogeneza. Kao i druge kronične komplikacije, dijabetička nefropatija povezana je s kroničnom hiperglikemijom. Točan mehanizam te povezanosti nije u potpunosti razjašnjen, ali smatra se da je uključeno više čimbenika: hemodinamske promjene bubrežne mikrocirkulacije (glomerularna hiperperfuzija i hiperfiltracija, povećani glomerularni kapilarni tlak), produkcija bioloških molekula (faktori rasta, angiotenzin II, endotelin) te strukturne promjene glomerula (povećanje ekstracelularnoga matriksa, zadebljanje glomerularne membrane, ekspanzija mezangija, fibroza). Prirodni tijek dijabetičke nefropatije u većoj je mjeri predvidiv u oboljelih od šećerne bolesti tip 1 nego kod oboljelih od šećerne bolesti tip 2. S obzirom na to da se dijabetička nefropatija razvija kod samo 20 do 40 % oboljelih, i drugi čimbenici moraju biti uključeni u razvoj bubrežnoga oštećenja. Ostali rizični čimbenici za razvoj dijabetičke nefropatije su: loša kontrola glikemije, duže trajanje šećerne bolesti, prisutnost drugih mikrovaskularnih komplikacija, etnička pripadnost (Azijci, Piama Indijanci), od ranije postojeća arterijska hipertenzija te obiteljska anamneza pozitivna na arterijsku hipertenziju ili dijabetičku nefropatiju.
Prirodni tijek dijabetičke nefropatije. Prve promjene kod oboljelih od šećerne bolesti tip 1 su glomerularna hiperperfuzija i glomerularna hipertrofija koje su povezane s pojačanom glomerularnom filtracijom i one se u većini slučajeva vide već kod postavljanja dijagnoze. Tijekom prvih pet godina bolesti dolazi do zadebljanja glomerularne membrane, progresije glomeluralne hipertrofije i povećanja mezangija uz razvoj arterioloskleroze, a glomerularna filtracija vraća se u normalu. U sljedećih 5 do 10 godina kod 40 do 50 % takvih bolesnika naći ćemo mikroalbuminuriju, a iako je ona važan rizičan čimbenik za razvoj makroalbuminurije, samo će se kod 50 % oboljelih ona i razviti u sljedećih deset godina. Kod manjega broja oboljelih od šećerne bolesti tip 1 kod kojih se registrira mikroalbuminurija ona kratko traje i spontano se povuče. Kada se jednom razvije makroalbuminurija, dolazi do postupnoga i progresivnog pada glomerularne filtracije te će se u sljedećih 7 do 10 godina razviti terminalni stadij kroničnoga bubrežnog zatajenja koji će zahtijevati neki od oblika nadomjesnoga liječenja bubrežne bolesti kod većine oboljelih. Nefropatija koja se razvija u oboljelih od šećerne bolesti tip 2 razlikuje se od one kod oboljelih od tip 1 na nekoliko načina: 1) mikroalbuminurija ili makroalbuminurija mogu biti prisutne već u trenutku dijagnoze, što je posljedica dugoga asimptomatskog perioda i kasnijega postavljanja dijagnoze; 2) hipertenzija češće komplicira mikroalbuminuriju ili makroalbuminuriju te 3) mikroalbuminurija je manje prediktivna za progresiju dijabetičke nefropatije, tj. napredovanja u makroalbuminuriju.
Patologija. Tri su glavne histopatološke promjene u dijabetičkoj nefropatiji: 1) povećanje (ekspanzija) mezangija; 2) zadebljanje glomerularne bazalne membrane te 3) glomerularna skleroza. Težina dijabetičke nefropatije procjenjuje se na osnovi debljine glomerularne bazalne membrane te volumena mezangija - što je membrana deblja i što je udio mezangija veći, to je i nefropatija teža. Uslijed hipertrofije glomerula dolazi i do povećanja bubrega u cijelosti, po čemu se dijabetička nefropatija razlikuje od drugih uzroka kroničnoga bubrežnog zatajenja kod kojih su bubrezi smanjenoga volumena.
Klinička slika. Dijabetička nefropatija dugo je asimptomatska bolest i najčešće se dijagnosticira u asimptomatskom periodu pri analizi jutarnjega uzorka urina ili analizi 24-satnoga urina na albumine i proteine (asimptomatska albuminurija ili proteinurija). S napredovanjem bolesti može se razviti i nefrotski sindrom. S postupnim padom glomerularne filtracije postupno se razvija i kronično bubrežno zatajenje.
Dijagnoza. Dijabetička nefropatija najčešće se dijagnosticira u asimptomatskoj fazi pretragom urina na albuminuriju. To se postiže pretragom urina jednom godišnje kod svih oboljelih od šećerne bolesti (probir na dijabetičku nefropatiju). Osim mikroalbuminurije, određuje se i glomerularna filtracija, prate se ureja i kreatinin. Po potrebi se preporuča ultrazvuk bubrega i procjenjuje potreba za biopsijom bubrega, osobito kod bolesnika koji se prezentiraju kao nefrotski sindrom nejasne etiologije.
Određivanje mikroalbuminurije. Mikroalbuminuriju se može određivati u 24-satnom urinu ili u jutarnjem uzorku urina. Ako se mjeri količina izlučenoga albumina u 24-satnom urinu, onda o albuminuriji govorimo ako je koncentracija albumina između 30 i 299 mg/dU. Kada se u urinu mjeri više od 300 mg/dU, govorimo o makroalbuminuriji, a kada je više od 3000 mg/dU, govorimo o nefrotskom sindromu. Ako mikroalbuminuriju procjenjujemo na osnovi jednoga (po mogućnosti prvoga jutarnjeg) uzorka urina, onda se procjenjuje omjer albumina i kreatinina iz uzorka urina, a omjer veći od 30 mg/g (ili > 0,03 mg/mg) govori u prilog postojanju mikroalbuminurije.
Procjena glomerularne filtracije vrši se na temelju određivanja klirensa kreatinina (KEK) koji pokazuje koliko bubreg kreatinina iz plazme pročisti u urin u jedinici vremena. Glomerularna filtracija odraz je funkcijskoga stanja bubrega, pa se smanjenje funkcije uslijed nefropatije odražava kao smanjena glomerularna filtracija, a time i smanjen klirens kreatinina.
Azotemija (povišene vrijednosti ureje i kreatinina) javlja se sa smanjenjem glomerularne filtracije, a najčešće se to događa kada se ona smanji ispod 25 - 30 %. Tada dolazi i do drugih laboratorijskih poremećaja vezanih uz kroničnu bubrežnu bolest.
Ultrazvuk bubrega pokazuje uvećane bubrege što je diferencijalno dijagnostički dobar pokazatelj, s obzirom na to da su kod drugih slučajeva kronične bubrežne bolesti prisutni smanjeni bubrezi. U slučaju nefrotskoga sindroma treba razmotriti i biopsiju bubrega.
Liječenje. Osnovni ciljevi liječenja dijabetičke nefropatije su: kontrola glikemije, kontrola arterijskoga tlaka, uvođenje ACE inhibitora ili ARB-ova u terapiju te kontrola lipidograma. Također, u slučaju razvoja dijabetičke nefropatije treba ograničiti unos proteina te prilagoditi terapiju šećerne bolesti s obzirom na bubrežnu funkciju. U terminalnoj fazi bolesti potrebno je provoditi klasično nadomjesno liječenje.
Kontrola glikemije jedna je od ključnih mjera u liječenju dijabetičke nefropatije. Poboljšanje glikemije odgađa pojavu mikroalbuminurije kod oboljelih od šećerne bolesti i tip 1 i tip 2, no nije jasno ima li poboljšana glikemija veći utjecaj na daljnju progresiju bubrežnoga oštećenja.
Kontrola arterijskoga tlaka također je bitan čimbenik u liječenju dijabetičke nefropatije, s obzirom na to da su brojne studije pokazale učinkovitost strože kontrole arterijskoga krvnog tlaka u smanjenju albuminurije i usporenju progresije bubrežene bolesti kod oboljelih od šećerne bolesti i tip 1 i tip 2. Kod takvih je bolesnika potrebno tlak održavati manjim od 130/80 mmHg.
Primjena ACE inhibitora ili inhibitora angiotenizin-II receptora (ARB) dokazano usporava progresiju mikroalbuminurije u makroalbuminuriju, a time i napredovanje bubrežne bolesti u oba tipa šećerne bolesti. Obje skupine lijekova imaju ekvivalentan učinak na poboljšanje bubrežne funkcije, stoga se i preporučuju kao lijek izbora u liječenju hipertenzije kod bolesnika sa šećernom bolesti. U slučaju nepodnošenja ta dva lijeka, kontrolu arterijskoga tlaka treba provoditi primjenom blokatora kalcijskih kanala, beta blokatora ili diuretika, no nije dokazan njihov povoljan učinak na samu albuminuriju. Treba uzeti u obzir da neki blokatori kalcijskih kanala mogu pogoršati albuminuriju.
Redukcija unosa proteina. Kod bolesnika s proteinurijom preporuča se redukcija unosa proteina u organizam. Bolesnicima s mikroalbuminurijom preporučena dnevna doza proteina iznosi 0,8 do 1,0 g/kg, a u slučaju makroalbuminurije trebala bi iznositi manje od 0,8 g/kg.
Prilagodba terapije šećerne bolesti. Kod bolesnika kod kojih se razvije bubrežno zatajanje treba prilagoditi samu terapiju šećerne bolesti: s progresijom bubrežnoga oštećenja potrebe za inzulinom su sve manje, s obzirom na to da je bubreg mjesto razgradnje inzulina. Metformin treba isključiti iz terapije kada dođe do pada vrijednosti glomerularne filtracije ispod 20 ml/min.
Liječenje uznapredovale faze bubrežnoga zatajenja. Kod uznapredovale faze dijabetičke nefropatije i razvoja terminalne faze bubrežnoga zatajenja pristupa se klasičnom nadomjesnom liječenju. Hemodijaliza ovih bolesnika udružena je s većom učestalošću komplikacija, kao što su hipotenzija (zbog pridružene autonomne neuropatije), otežan vaskularni pristup, brži razvoj retinopatije te akcelerirana ateroskleroza.
DIJABETIČKA NEUROPATIJA
Definicija. Dijabetička neuropatija jedna je od najčešćih kroničnih komplikacija šećerne bolesti koja zahvaća više od 50 % starijih osoba oboljelih od šećerne bolesti tip 1 i tip 2. Razlikuju se periferna i autonomna dijabetička neuropatija. Periferna dijabetička neuropatija može zahvatiti više živaca, pa govorimo o polineuropatiji, ili može biti ograničena na jedan živac, pa govorimo o mononeuropatiji. Najčešći oblici periferne polineuropatije su distalna simetrična polineuropatija te proksimalna motorna polineuropatija.
Etiopatoegenza i patološka obilježja. Kao i kod drugih kroničnih komplikacija, glavni etiopatogenetski čimbenik je kronična hiperglikemija. Ostali čimbenici rizika su povećan indeks tjelesne mase, pušenje, hipertenzija, dislipidemija i pozitivna anamneza kardiovaskularnih događaja. Glavna histopatološka obilježja zahvaćenih živčanih vlakana su: 1) aksonalna degeneracija koja označava gubitak mijelinizirajućih i nemijelinizirajućih vlakana; 2) zadebljanje bazalne membrane Schwannovih stanica; 3) segmentalna demijelinizacija te 4) zadebljanje bazalnih membrana intraneuralnih kapilara s nastankom mikrotromba.
Distalna simetrična senzomotorna polineuropatija najčešći je oblik dijabetičke neuropatije. Iako navedenim promjenama mogu biti zahvaćeni bilo koji somatski živci, najčešće su zahvaćeni distalni senzomotorni živci nogu te u manjoj mjeri ruku. Dominantno patološki proces zahvaća osjetna vlakna i to i mala nemijalizirana C-vlakna (prijenos osjeta bola i temperature), kao i velika mijelizirana vlakna (prijenos osjeta dodira, vibracije, propriocepcije). Motorna vlakna izuzetno su rijetko zahvaćena. U početnoj su fazi bolesnici asimptomatski, a ispadi se uočavaju tek neurološkim pregledom (gubitak osjeta vibracije, temperature, finoga osjeta i sl.). S vremenom se razvijaju tipični simptomi kao što su utrnulost, pojava trnaca, osjećaj žarenja, a bolesnici se žale na bol u donjim ekstremitetima. Bol se javlja u mirovanju, a karakteristična je tendencija pogoršanja bola tijekom noći. S napredovanjem bolesti bol postupno iščezava, ali ostaje senzorni deficit. Kod manjega se broja bolesnika uslijed zahvaćenosti motornih vlakna javlja i specifičan motorički deficit.
Proksimalna motorna neuropatija ili dijabetička amiotrofija javlja se poglavito kod starijih osoba sa šećernom bolesti tip 2, i to češće u muškaraca. Radi se o teškoj i progresivnoj bolesti koja nastaje uslijed promjena na motornim živčanim vlaknima koji inerviraju proksimalne mišiće donjih udova i zdjelice, a rjeđe se radi o zahvaćenosti motornih živčanih vlakana za mišiće ramena i nadlaktice. Bolest se manifestira bolovima u bedrima, gluteusima i zdjelici, nakon čega slijedi najprije slabost, a zatim i atrofija mišića zdjelice i bedra (najčešće iliopsoas, obturator, aduktori zdjelice). Uz navedene simptome, bolest može biti praćena značajnijim gubitkom na tjelesnoj težini, što se naziva neuropatskom kaheksijom. Takvi bolesnici često ne mogu obavljati niti osnovne svakodnevne aktivnosti. Iako se kod većine bolesnika dogodi oporavak neurološkoga deficita u periodu od 12 mjeseci, neki deficiti mogu trajno ostati.
Periferna dijabetička mononeuropatija označava promjene koje uključuju jedan živac, bilo periferni, bilo kranijalni. Karakteristično je da se promjene i simptomi razvijaju brzo, ali se brzo i oporavljaju. Najčešće su zahvaćeni III. i VI. kranijalni živac (što rezultira pojavom dvoslika) te femoralni živac s dominantnim motoričkim ispadima. Rijetko su zahvaćeni drugi živci. Oporavak deficita očekuje se kroz 6 do 12 mjeseci.
Autonomna neuropatija označava promjene na živčanim vlaknima autonomnoga živčanog sustava (kolinergična i adrenergična vlakna), a javlja se kod bolesnika sa šećernom bolesti koja traje duže vrijeme. Ove promjene mogu imati utjecaj na brojne organe i organske sustave, što se može manifestirati različitom simptomatologijom. Studije su pokazale manju povezanost između loše regulacije glikemije s pojavnošću autonomne neuropatije (za razliku od periferne), ali bolja regulacija glikemije nije povezana s poboljšanjem simptoma. S obzirom na to da autonomna dijabetička neuropatija može zahvatiti brojne organe i organske sustave, simptomi su različiti: kardiovaskularni (postularna hipotenzija, poremećaji srčanoga ritma); gastrointestinalni (disfagija, gastropareza, noćna dijareja s ili bez fekalne inkontinencije, konstipacija); genitourinarni (urinarna inkontinencija, otežana mikcija, atonija mokraćnoga mjehura, erektilna disfunkcija, retrogradna ejakulacija), vazomotorni (hladne okrajine, lokalni edemi) te ostali (anhidroza, hiperhidroza, poremećaji pupilarne motorike).
Dijagnostička obrada i evaluacija u domeni su specijalista neurologije. Dijagnoza se postavlja na temelju anamnestičkih i kliničkih podataka, somatskoga i neurološkog pregleda (ispitivanje osjetnoga i motoričkog deficita) te elektrofiziološkom obradom – elektromioneurografija. Potrebno je isključiti i druge moguće uzroke neuropatija (diferencijalna dijagnoza).
Liječenje dijabetičke neuropatije nije zadovoljavajuće. Terapijske mjere uključuju kontrolu glikemije, arterijskoga tlaka i dislipidemije, uz izbjegavanje neurotoksina (pušenje) te primjenu vitamina B skupine u svrhu neuroprotekcije. Neuropatski bol teško je kupirati analgeticima, ali učinak je bolji ako im se dodaju antidepresivi kao pomoćni lijekovi (triciklički antidepresivi, selekivni inhibitori ponovne pohrane serotonina), a nekada se uvode i antikonvulzivi kao pomoćni lijekovi (gabapentin, karbamazepin, lamotrigin). U liječenju motornih i osjetnih deficita veliku ulogu imaju metode fizikalne medicine. Liječenje autonomne dijabetičke neuropatije temelji se na simptomatskim mjerama: postularna hipotenzija (fludrokortizon, agonist α receptora, nesteroidni protuupalni lijekovi), gastropareza (metoklopramid, eritromicin), dijareja (loperamid, okteroitid, antibiotici širokoga spektra), konstipacija (laksativi, prehrana s puno ostataka, tjelovježba), atonija mjehura (povremene samokateterizacije), hiperhidroza (antikolinergički lijekovi, klonidin), erektilna disfunkcija (sildenafil, alprostadil) i dr.
DIJABETIČKO STOPALO
Definicija. Dijabetičko stopalo poseban je klinički entitet koji se često javlja kod bolesnika s loše reguliranom šećernom bolesti, a uključuje sporo cijeljenje plantarnih ulceracija koje nastaju kao rezultat beznačajnih oštećenja (lagani udarac, neodgovarajuća obuća i sl.). Ako se na vrijeme ne prepozna i ne liječi, moguć je razvoj teških komplikacija kao što su celulitis, apsces te osteomijelitis.
Etiopatogeneza. Promjene nastaju kao rezultat traume (obično beznačajne) u prisutnosti drugih patofizioloških čimbenika. Glavni patofiziološki čimbenici razvoja dijabetičkoga stopala su: 1) senzomotorna neuropatija (umanjen osjet bola, abnormalnosti propriocepcije), 2) periferna vaskularna bolest (hipoperfuzija kože i mišića stopala), 3) autonomna neuropatija (poremećeni autoregulacijski mehanizmi mikrocirkulacije) te 4) oslabljen imunološki odgovor (otežana borba s infekcijama).
Klinička slika. Promjene mogu nastati na bilo kojemu dijelu stopala, a nastaju kao rezultat beznačajne traume (manji udarac, pritisak od neodgovarajuće obuće, ozljeda pri rezanju noktiju i sl.). S obzirom na to da je kod ovih bolesnika uglavnom razvijena senzorna neuropatija, bolesnici nemaju osjet neugode ili bola što je preduvjet za dulje izlaganje traumi. Radi se obično o malim, plitkim ulceracijama koje se sekundarno inficiraju pa postaju crvene, razvija se oteklina, a često i gnojna sekrecija. S obzirom na postojeću imunonedostatnost, često se upalni proces širi u okolno rahlo tkivo (celulitis) ili se formiraju gnojne kolekcije (apsces), a moguć je i prodor u koštano-zglobne strukture (osteomijelitis). Uz upalne promjene, dolazi i do progresije arterijske insuficijencije, pa se u konačnici razvijaju nekroza i gangrena te se mora pristupiti amputaciji. Moguć je i prodor uzročnika u krv, uz nastanak sepse koja dodatno ugrožava bolesnike sa šećernom bolesti.
Dijagnoza se postavlja na osnovi kliničkoga (lokalnog) nalaza, uz ostale anamnestičke podatke (trajanje šećerne bolesti, regulacija glikemije, postojanje drugih kroničnih komplikacija i sl.). Potrebno je uzeti i bris rane kako bi se obavila mikrobiološka dijagnostika i započelo liječenje odgovarajućim antibiotikom prema antibiogramu. U svrhu procjene arterijske irigacije, može se obaviti i angiografija.
Liječenje. Jednom nastale promjene na stopalu potrebno je što ranije i što agresivnije liječiti kako bi se spriječio razvoj komplikacija. Potrebno je redovno provoditi lokalno previjanje rane i uklanjanje nekrotične mase (nekrektomija), što je u nadležnosti kirurga, ordinirati antibiotike prema antibiogramu te osigurati prikladnu kontrolu glikemije. Lokalna primjena rekombinantnoga humanog faktora rasta iz trombocita može umjereno poboljšati cijeljenje rane. U slučaju uznapredovalih promjena te ako provedene mjere ne daju rezultate, mora se pristupiti kirurškoj amputaciji.
Prevencija je najbitnija mjera u sprečavanju nastanka dijabetičkog stopala. Mjere prevencije uključuju bolju kontrolu glikemije, eliminaciju čimbenika rizika (npr. pušenje, smanjenje tjelesne mase), edukaciju bolesnika (svakodnevni pregled stopala, održavanje higijene kože stopala, anatomska obuća, izbjegavanje hodanja bosih nogu) te redovite liječničke preglede.
OSTALE KRONIČNE KOMPLIKACIJE ŠEĆERNE BOLESTI
Kardiovaskularne komplikacije. Učestalost kardiovaskularnih oboljenja povećana je kod oboljelih od šećerne bolesti i tip 1 i tip 2, što su pokazale različite studije. One nastaju kao posljedica ubrzanoga razvoja ateroskleroze uslijed sinergističkoga učinka hiperglikemije i ostalih kardiovaskularnih rizika (dislipidemija, hipertenzija, debljina, smanjena fizička aktivnost, pušenje). Dodatni rizični čimbenici za obolijevanje od kardiovaskularnih bolesti kod ovih su bolesnika mikroalbuminurija, makroalbuminurija, povišenje koncentracije kreatinina u serumu, abnormalna funkcija trombocita te inzulinska rezistencija i hiperinzulinemija. Kod bolesnika s inzulinskom rezistencijom nalazimo povišene vrijednosti inhibitora aktivacije plazminogena (PAI-1) te fibrinogena što potiče koagulacijsku kaskadu, a slabi fibrinolitičku ulogu, što sve pridonosi trombotskim incidentima. Dodatno rizik povećava disfunkcija endotelnih stanica. Najčešće kardiovaskularne komplikacije su koronarna bolest, cerebrovaskularna bolest te bolest perifernih arterija. Klinički pristup, odnosno dijagnostika i liječenje ovih bolesnika, individualan je. Poboljšana kontrola glikemije te liječenje i uklanjanje ostalih kardiovaskularnih čimbenika rizika (arterijska hipertenzija, dislipidemija) imaju pozitivan učinak na kardiovaskularni morbiditet i mortalitet.
Gastrointestinalne komplikacije posljedica su prvenstveno autonomne neuropatije. Najčešći poremećaji gastrointestinalnoga sustava povezani sa šećernom bolesti su gastropareza (atonija i usporeno praženje želuca) te abnormalnosti motiliteta tankoga i debelog crijeva, što se očituje kao dijareja ili konstipacija. Gastropareza je obilježena gubitkom teka, ranom sitošću, mučninom i povraćanjem te nadutošću. Može ju se dokazati scintigrafijom radioobilježenoga obroka, čime se može dokumentirati usporeno pražnjenje želuca, iako dobiveni nalazi nisu u korelaciji sa simptomima bolesti. Poremećaj crijevnoga motiliteta klinički je obilježen noćnim proljevima te naizmjeničnim epizodama opstipacije. Moguća je i disfunkcija jednjaka, ali ona uglavnom ostaje asimptomatska. Liječenje podrazumijeva kontrolu glikemije te primjenu simptomatskih lijekova. Kod gastropareze pomaže primjena metoklopramida (5 - 10 mg) ili dompridona (10 - 20 mg) prije svakoga obroka. Eritromicin interferirajući s motilin receptorima može poticati pražnjenje želuca. U slučaju upornih proljeva može se primijeniti loperamid ili, ako ne izaziva reakciju, i oktreotid. Kod sumnje na sindrom bakterijskoga prerastanja, indicirana je primjena antibiotika širokoga spektra. U liječenju konstipacije savjetuje se hrana s puno ostataka, dostatna hidracija, tjelovježba i eventualno primjena laksativa (lokalni glicerinski čepići i sl.).
Genitourinarne komplikacije vezane uz dugotrajnu šećernu bolest uključuju u prvom redu disfunkciju mokraćnoga mjehura (dijabetička cistopatija) i erektilnu disfunkciju. Dijabetička cistopatija uzrokovana je poremećajima inervacije uslijed razvijene neuropatije. Simptomi uključuju izostanak osjećaja punoće mjehura, nemogućnost potpunoga pražnjenja mjehura, smanjenu učestalost mokrenja te učestale infekcije. Dijagnoza se postavlja cistometrijski, a liječenje se provodi ponavljanim samokateterizacijama, a može se pokušati i s primjenom betanekola u dozi od 10 - 50 mg tri puta na dan. Erektilna disfunkcija posljedica je neuropatskih, vaskularnih i psiholoških čimbenika, a najčešće se liječi medikamentozno (sildenafil 50 - 100 mg sat vremena prije spolne aktivnosti). Mogu se primjenjivati i drugi lijekovi, kao što su vardenafil ili tadalafil. Potreban je poseban oprez kod bolesnika s kardiovaskularnim rizikom (npr. kardiovaskularni incident u proteklih šest mjesci, nekontrolirane aritmije, izražene hipertenzije ili hipotenzije).
Mukokutane komplikacije. Kod bolesnika sa šećernom bolesti cijeljenje rana otežano je i produljeno. Od ostalih specifičnih kožnih promjena koje se povezuje s dugotrajnom šećernom bolesti tu su dijabetička dermatopatija i necrobiosis lipoidica diabeticorum. Dijabetička dermatopatija (engl. "shin spots") označava promjene na pretibijalnoj regiji koje počinju kao eritem koji se razvija u hiperpigmentaciju, a javlja se kao odgovor na minornu mehaničku traumu. Necrobiosis lipoidica diabeticorum rijetka je kožna promjena nerazjašnjenoga mehanizma koju uglavnom susrećemo kod mlađih bolesnika sa šećernom bolesti tip 1. Obično počinje kao eritematozni plak u pretibijalnoj regiji koji se postupno širi, tamni, poprima nepravilne rubove uz centralnu atrofiju i razvoj ulceracije. Promjena je obično bolna. Liječi se topičkom primjenom glukokortikoida. Kod bolesnika sa šećernom bolesti tip 1 češća je i pojava vitiliga i drugih dermatoza, kao što je crna akantoza, lokalizirani anularni granulom te sklerodermija. Lipoatrofija i lipodistrofija povezane su s primjenom inzulinske terapije u supkutano masno tkivo. Češća je i pojava mukokutanih gljivičnih infekcija, osobito kandidijaze.
Koštano-zglobne komplikacije. Bolesnici sa šećernom bolesti mogu imati sklonost određenim koštano-mišićnim oboljenjima. Uslijed glikozilacije kolagena i drugih proteina vezivnoga tkiva, kod ovih je bolesnika veća učestalost razvoja sindroma karpalnoga tunela, sindroma smrznutoga ramena (adhezivni kapsulitis) te Dupuytrenove kontrakture. Žene sa šećernom bolesti tip 1 imaju veći rizik od razvoja patoloških fraktura u usporedbi sa ženama koje nemaju šećernu bolest, no studije o gustoći koštane mase i riziku od fraktura kod bolesnika sa šećernom bolesti i dalje su kontradiktorne. Difuzna idiopatska hiperostoza skeleta karakterizirana je osifikacijom prednjega longitudinalnog ligamenta kralježnice što dovodi do njezine krutosti i ograničenja pokretljivosti. Osim šećerne bolesti kao glavnoga rizičnog čimbenika razvoja toga rijetkog stanja, spominju se i debljina, hipertenzija te displipidemija.
Infekcije. Bolesnici sa šećernom bolesti imaju veću učestalost infekcija, a to je posljedica promjena u stanično posredovanoj imunosti te abnormalnoj funkciji fagocita povezanih s hiperglikemijom. Osim veće učestalosti uobičajenih infekcija, kod osoba sa šećernom bolesti javljaju se i infekcije koje nisu karakteristične kod ostalih osoba, a u tu skupinu ubrajamo emfimatoznu upalu žučnoga mjehura i mokraćnoga mjehura, rinocerebralnu mukormikozu te invazivnu infekciju vanjskoga uha (Pseudomonas aeruginosa). Učestalost pneumonija, urinarnih i kožnih infekcija veća je nego u populaciji osoba bez šećerne bolesti. Uzročnici tih infekcija slični su kao i u drugim populacijama, s time da su kod bolesnika sa šećernom bolesti nešto češći uzročnici gram-negativne bakterije te S. aureus. Karakteristično, uslijed glikozurije kod bolesnika sa šećernom bolesti nalazimo veću učestalost genito-urinarnih gljivičnih infekcija.
POSEBNA STANJA VEZANA UZ ŠEĆERNU BOLEST
LIJEČENJE ŠEĆERNE BOLESTI U BOLNIČKIM UVJETIMA
Mnogi bolesnici sa šećernom bolesti bit će tijekom života hospitalizirani iz različitih razloga, a to može imati utjecaja na samu terapiju šećerne bolesti. Smatra se da oko 10 - 15 % svih hospitaliziranih osoba ima šećernu bolest, a da 30 % hospitaliziranih osoba sa šećernom bolesti ima neodgovarajuću regulaciju glikemije tijekom hospitalizacije što proizlazi iz više razloga (bolesnici nemaju uobičajenu prehranu kao u kućnim uvjetima, različite pretrage zahtijevaju da bolesnici budu natašte i sl.)
Kod bolesnika koji mogu normalno uzimati hranu nastoji se nastaviti s uobičajenim izvanbolničkim načinom prehrane te se primjenjuje terapija šećerne bolesti jednaka kao i u kućnim uvjetima, uz eventualne korekcije doze inzulina ovisno o nalazu dnevnoga profila glukoze, ako su već od ranije na inzulinskoj terapiji. Ako se to ne može postići, preporuča se primjena inzulinske terapije i to najčešće u obliku jedne večernje doze dugodjelujućega inzulinskog analoga s ili bez primjene prandijalnih inzulina.
Regulacija glikemije bolesnicima koji se podvrgavaju kirurškim zahvatima dodatan je izazov jer fiziološki stres operacije dodatno pogoršava hiperglikemiju (pojačana produkcija kontraregulatornih inzulinskih hormona kao što su katekolamini, hormon rasta, kortizol i dr.). Bolesnike treba hospitalizirati jedan do tri dana prije zahvata i regulirati glikemiju tako da ona prije zahvata iznosi 7 do 9 mmol/L.
Za bolesnike koji glikemiju kontroliraju isključivo dijabetičkom prehranom nema značajnijih preporuka, osim ako će glikemija biti znatnije narušena samim zahvatom (veće operacije, duži oporavak i sl.). Takvim se bolesnicima preporuča privremena supkutana primjena brzodjelujućih inzulinskih preparata (humani inzulin, brzodjelujući inzulinski analozi).
Bolesnici koji uzimaju oralne antihiperglikemike trebaju ih izostaviti na dan operacije (metformin se treba izostaviti tri dana prije operativnoga zahvata). U slučaju značajne hiperglikemije primjenjuju se brzodjelujući inzulini kao privremeno rješenje. U slučaju da se tako ne može osigurati odgovarajuća kontrola glikemije, indicirana je primjena infuzije inzulina prema shemi opisanoj u nastavku. Čim bolesnik nakon operacije počne normalno jesti, potrebno je iznova uvesti oralne hipoglikemike. Preporuča se prije uvođenja oralnih antihiperglikemika (osobito metformina) postoperativno provjeriti koncentraciju kreatinina u serumu kako bi se ustvrdilo da nije došlo do pogoršanja bubrežne funkcije, što bi dovelo do neželjenih učinaka nakon ponovnoga uvođenja oralnih hipoglikemika.
Kod bolesnika koji od ranije liječe šećernu bolest inzulinskom terapijom potrebna je korekcija terapije za vrijeme operativnoga zahvata. Postoje standardizirani protokoli i preporuke koje uključuju dva glavna čimbenika, a to su tip šećerne bolesti (tip 1 ili tip 2) te opseg operativnoga zahvata: manji kirurški postupak (traje manje od 2 sata, a odmah nakon njega moguća je prehrana) te veći kirurški postupak (traje dulje od 2 sata, uključuje otvaranje tjelesnih šupljina, bolesnici nakon zahvata ne mogu jesti). U Tablici 8.10. su preporuke za inzulinsko liječenje tijekom kirurškoga tretmana.
|
Tablica 8.10. Preporuke za inzulinsko liječenje tijekom kirurškoga zahvata
|
|
Tip šećerne bolesti
|
Manji kiruški postupak
|
Veći kirurški postupak
|
|
Tip 2 (liječenje predmiješanim inzulinima ili bazal-bolus shema)
|
Izostaviti inzulinsku terapiju na da operacije. Uključiti infuziju 5 %-tne glukoze te mjeriti glukozu uz primjenu brzodjelujućih inzulina supkutano svakih 4 do 6 sati.
|
Primjenjuje se isti protokol kao kod manjega kirurškog postupka, no ako se može postići zadovoljavajuća kontrola glikemije, primjenjuje se infuzija inzulina.
|
|
Tip 1 (liječenje inzulinskom pumpom, bazal-bolus shema)
|
Pacijentima na terapiji inzulinskom pumpom prekida se primjena inzulina večer prije operacije kada se daje jedna doza dugodjelujućega inzulinskog analoga. Na dan operacije uključuje se infuzija 5 %-tne glukoze, mjeri se glukoza te se daju brzodjelujući inzulini svakih 4 do 6 sati.
|
Na dan operacije bolesniku se uključuje infuzija inzulina te se primjenjuje dok bolesnik ne počne jesti nakon operacije, kada se vraća na prethodni način liječenja.
|
Doziranje brzodjelujućih inzulinskih analoga u smislu korekcije glikemije kod bolesnika koji ne jedu izvodi se svakih 4 do 6 sati nakon mjerenja glukoze u plazmi. Kod mršavih se osoba doza brzodjelujućega inzulinskog analoga koji želimo primijeniti računa tako da za svakih 2,7 mmol/L glukoze iznad ciljne vrijednosti primijenimo 1 ij inzulina, a kod pretilih se osoba za svakih 2,7 mmol/L glukoze iznad ciljane vrijednosti primjenjuje 2 ij inzulina.
Infuzije inzulina primjenjuju se kada se ne može održati normoglikemija na drugi način. Postoji više shema za primjenu infuzije inzulina, a najraširenija je takozvana glukoza-inzulin-kalij shema (GIK shema) koja podrazumijeva pripremu 1000 ml 10 %-tne glukoze u koju se dodaje 16 jedinica humanoga (regularnog) inzulina te 20 mmol KCl-a, a primjenjuje se brzinom od 100 ml/h. Tijekom primjene je potrebno kontrolirati razinu glikemije i kalija te po potrebi korigirati terapiju. Alternativna je metoda odvojena primjena glukoze i inzulina putem inzulinskih pumpi. Bolesnicima s jetrenom bolesti, pretilim bolesnicima te bolesnicima na kortikosteroidnoj terapiji potrebne su veće doze inzulina. Infuzije inzulina primjenjuju se sve dok bolesnici ne počnu ponovno jesti, kada se vraćaju na prijašnji režim liječenja šećerne bolesti. Infuzija inzulina prekida se 30 minuta nakon prve supkutane primjene inzulina. S obzirom na to da su nekoliko dana nakon operacije potrebe za inzulinom varijabilne, preporuča se višednevna primjena brzodjelujućih inzulina uz jednu večernju dozu dugodjelujućega inzulinskog analoga.
Kontrola glikemije u jedinicama intenzivnoga liječenja kod kritično oboljelih osoba ne bi trebala biti stroga, a ciljne vrijedosti glikemije kod tih su bolesnika 7,8 do 10,0 mmol/L. Velika randomizirana klinička studija NICE-SUGAR pokazala je povećanu smrtnost i veću učestalost epizoda teških hipoglikemija kod bolesnika liječenih u jedinicama intenzivnoga liječenja (mnogi od tih bolesnika bili su mehanički ventilirani) kod kojih se težilo strogoj regulaciji glikemije (4,5 – 6,0 mmol/L), za razliku od skupine bolesnika kod kojih je provođena manje stroga regulacija glikemije i ona je iznosila oko 8 mmol/L. I druge slične studije pokazale su da stroga kontrola glikemije kod kritično oboljelih bolesnika nosi rizik od teških hipoglikemija i veće smrtnosti. Kod bolesnika u jedinicama intenzivnoga liječenja preporučuje se primjena infuzije inzulina ili primjena inzulina preko infuzijske pumpe, s obzirom na to da je supkutana apsorpcija inzulina kod teško oboljelih osoba nepredvidiva. Kod primjene infuzije inzulina potrebne su češće kontrole glukoze i kalija u plazmi te njihova korekcija ako je to potrebno. S obzirom na kratak poluživot intravenski primijenjenoga inzulina, prije prekida infuzije inzulina potrebno je supkutano primijeniti dugodjelujući inzulinski analog kako bi se izbjegla epizoda bez inzulina. On se primjenjuje 2 do 4 sata prije prekida intravenskoga unosa inzulina.
Bolesnici koji su na totalnoj parenteralnoj prehrani imaju povećane potrebe za inzulinom, a često i bolesnici koji prije nisu imali šećernu bolest postaju hiperglikemični i zahtijevaju primjenu inzulina. Ako se radi o kritično oboljelim osobama u jedinicima intenzivnoga liječenja, potrebna je primjena intravenskoga inzulina (infuzijom ili perfuzorom) uz češće kontrole glikemije i prilagodbu doze.
ŠEĆERNA BOLEST I AKUTNI INFARKT MIOKARDA
Hiperglikemija je često prisutna kod bolesnika s akutnim infarktom miokarda. Kod nekih se radi o stresnoj hiperglikemiji, kod nekih o ranije postojećoj, nedijagnosticiranoj šećernoj bolesti, a kod nekih o pogoršanju glikemije u ranije dijagnosticiranoj šećernoj bolesti. Hiperglikemija tijekom akutnoga infarkta miokarda povećava stopu smrtnosti. Preporuča se u periinfarktnom periodu glikemiju regulirati inzulinskom terapijom, a ako su bolesnici i uzimali oralne hipoglikemike, treba ih izostaviti u tom periodu. Studije su pokazale da je kontrola glikemije inzulinom kod bolesnika sa šećernom bolesti tip 2 pri akutnom infarktu miokarda povezana s dugoročnim smanjenjem mortaliteta od koronarne bolesti.
Preporuke za smanjenje mortaliteta od akutnoga infarkta miokarda kod bolesnika sa šećernom bolesti uključuju: 1) primarnu prevenciju od infarkta miokarda (stroga kontrola glikemije, arterijskoga tlaka i lipidograma); 2) odgovarajuće postupke u samom infarktu miokarda (primarna perkutana koronarna intervencija ili fibrinoliza, aspirin i klopidogrel, ACE inhibitor, beta blokator, inzulinska regulacija glikemije) te 3) sekundarnu prevenciju akutnoga infarkta miokarda (aspirin, beta blokator, ACE inhibitor, statini, stroga kontrola glikemije).
GESTACIJSKI DIJABETES
Definicija. Gestacijski dijabetes označava šećernu bolest koja se prvi puta dijagnosticira u trudnoći. Najčešće se razvija u drugom tromjesječju kada je i najjače izražena inzulinska rezistencija vezana uz trudnoću. Učestalost je različita i ovisi o rasnoj i etničkoj skupini, tako da se kreće u rasponu od 2 do 8 %. Radi se o kliničkom stanju koji nosi određene zdravstvene rizike i za majku i za čedo.
Etiopatogeneza. Kako je rečeno, trudnoća je povezana s inzulinskom rezistencijom uvjetovanom raznim metaboličkim promjenama. U normalnoj situaciji žene na to odgovaraju povećanim stvaranjem inzulina, tako da se ne razvija hiperglikemija. Kod nekih žena taj kompenzatorni mehanizam ne može održati euglikemiju te se kod njih razvija gestacijski dijabetes. Rizični su čimbenici za razvoj gestacijskoga dijabetesa: 1) opterećena obiteljska anamneza na šećernu bolest (1. koljeno); 2) indeks tjelesne mase veći od 30; 3) trudnica u dobi višoj od 30 godina; 4) porođajna masa djeteta iz prethodne trudnoće veća od 4110 g; 5) oštećena tolerancija glukoze u osobnoj anamnezi; 6) perinatalni gubitak ili malformacije u prethodnim trudnoćama; 7) majčina porođajna masa veća od 4100 g ili manja od 2700 g; 8) glikozurija na prvom pregledu; 9) sindrom policističnih jajnika; 10) terapija kortikosteroidima te 11) kronična/graviditetna hipertenzija.
Rizici za čedo. Rizici za čedo su mnogobrojni: 1) makrosomija (otežan porod, češće operativno dovršenje trudnoće), 2) fetalna organomegalija (hepatomegalija, kardiomegalija), 3) povećan neonatalni morbiditet (hipoglikemija, hiperbilirubinemija, hipokalcemija) te 4) povećan dugoročni morbiditet (pretilost, metabolički sindrom, šećerna bolest).
Rizici za majku. Iako se kod više od 90 % trudnica postiže spontana normoglikemija nakon porođaja, ostaje znatan zdravstveni rizik: 1) rizici vezani uz same komplikacije vezane uz porod; 2) gestacijski dijabetes u predstojećim trudnoćama; 3) razvoj šećerne bolesti kasnije u životu; 4) povećan kardiovaskularni rizik te 5) povećan rizik od nastanka metaboličkoga sindroma.
Dijagnoza. Danas postoje različiti kriteriji za dijagnozu gestacijskoga dijabetesa. Najrasprostranjeniji su kriteriji Svjetske zdravstvene organizacije i tzv. HAPO kriteriji (prema HAPO studiji). Kriteriji Svjetske zdravstvene organizacije podrazumijevaju GUP natašte veći od 7,0 mmol/L ili veći od 7,8 mmol/L u 120. minuti nakon oralnog unosa 75 g glukoze (OGTT). Ako je GUP natašte viši od 7,8 mmol/L, smatra se da je žena prije trudnoće imala razvijenu šećernu bolest, samo nije bila otkrivena. Takve žene ne zadovoljavaju kriterije gestacijskoga dijabetesa. HAPO kriteriji podrazumijevaju da jedna od sljedećih koncentracija glukoze u plazmi bude ista ili veća od graničnih vrijednsti: glukoza viša od 5,1 mmol/L natašte te glukoza viša od 10 mmol/L nakon jednog sata ili viša od 8,5 mmol/L nakon dva sata u oGTT testu sa 75 grama glukoze.
Sve žene s dijagnosticiranim gestacijskim dijabetesom moraju biti pod strogim nadzorom ginekologa koji trudnoću vodi kao ugroženu.
Liječenje uključuje pravilnu prehranu (dijabetička dijeta od 1800 kcal koja uključuje tri glavna obroka i tri međuobroka) uz odgovarajuću tjelovježbu, a ako se ne postigne zadovoljavajuća kontrola glikemije, u terapiju se uključuju inzulini (dugodjelujući inzulinski analozi kao bazalni inzulin te brzodjelujući inzulinski analozi kao prandijalni inzulini).
Preporuke za žene s gestacijskim dijabetesom nakon porođaja uključuju: 1) OGTT nakon 6 do 12 tjedana od poroda, 2) godišnju endokrinološku evaluaciju, 3) smanjenje tjelesne mase, 4) redovitu fizičku aktivnost te 5) prestanak pušenja.
TRUDNOĆA I PREEGZISTIRAJUĆA ŠEĆERNA BOLEST
Žene sa šećernom bolesti trebale bi proći prekoncepcijsku skrb prije nego ostanu trudne, a za vrijeme trudnoće trebaju biti češće kontrolirane s obzirom na povećane zdravstvene rizike i za majku i za čedo. U ranoj je trudnoći loše regulirana glikemija povezana s većom učestalošću spontanih pobačaja i kongenitalnih malformacija. U kasnoj je trudnoći loše regulirana glikemija povezana s većom učestalošću polihidramiona, makrosomije i komplikacija vezanih uz sam porod.
Kod većine žena sa šećernom bolesti tijekom trudnoće dolazi do pogoršanja i ubrzanja nastanka kroničnih komplikacija. Često se bilježi nastanak retinopatije ili pogoršanje nalaza ranije postojeće retinopatije. Također, ako je prije trudnoće bila prisutna mikroalbuminurija, ona češće prelazi u makroalbuminuriju, što može predisponirati pojavu preeklampsije i eklampsije. Često se bilježi i pogoršanje simptoma dijabetičke gastopareze što dovodi do upornih mučnina i povraćanja.
Iako su neki oralni antihiperglikemici proglašeni sigurnima za korištenje u trudnoći, današnje preporuke daju prednost inzulinima kao metodi kontrole glikemije u trudnoći i to po shemi bazal-bolus. Najčešće preporuke za primjenu brzodjelujućih inzulinskih analoga kao prandijalnih inzulina odnose se na primjenu inzulina asparta i inzulina lispro. NPH inzulin preporuča se za pokrivanje bazalnih potreba za inzulinom, a mogu se korisiti i inzulin glargin i degludek.
Definicija. Hipoglikemija je stanje snižene koncentracije glukoze u plazmi, a najbolje je definirana Whippleovim trijasom kojega čine: 1) simptomi povezani s hipoglikemijom; 2) niska koncentracija glukoze u plazmi mjerena pouzdanom metodom te 3) povlačenje simptoma nakon povećanja koncentracije glukoze u plazmi. Najnižom normalnom vrijednošću glukoze u plazmi smatra se vrijednost od 3,9 mmol/L, ali i niže se vrijednosti mogu fiziološki javiti u nekim stanjima, kao što je razdoblje kasno nakon obroka, tijekom trudnoće, te tijekom produljenoga gladovanja. Hipoglikemija je najčešće povezana s liječenjem šećerne bolesti, ali može se javiti i neovisno o njoj. Hipoglikemija je povezana s brojnim komorbiditetima, a ako dulje traje, može biti i fatalna.
Fiziologija. Glukoza je obligatan izvor energije za neurone, s obzirom na to da oni u fiziološkim uvjetima ne mogu sintetizirati glukozu de novo niti je pohraniti u obliku glikogena, pa je za normalnu funkciju neurona potrebna kontinuirana opskrba glukozom iz arterijske cirkulacije. Prema tome, pad razine glukoze u plazmi dovodi do poremećaja metabolizma i funkcioniranja neurona, a kako bi se spriječila takva događanja i velika kolebanja vrijednosti glukoze u plazmi, postoje mehanizmi koji se aktiviraju kada dođe do hipoglikemije i koncentraciju glukoze u plazmi vraćaju u fiziološki okvir.
Fiziološki, vrijednosti glukoze u plazmi natašte kreću se između 3,9 i 6,1 mmol/L s fiziološkim varijacijama kao što je npr. tranzitorno povišenje koncentracije glukoze u plazmi nakon obroka. Fiziološke vrijednosti glukoze u plazmi između obroka održavaju se uslijed endogene sinteze glukoze (glukoneogeneza u jetri i bubrezima) te razgradnjom glikogena (glikogenoliza) u jetri. Jetrene zalihe glikogena dostatne su za održavanje normoglikemije tijekom prosječno 8 sati gladovanja, iako taj period može biti i kraći ako su potrebe za glukozom veće (pojačan rad, bolest). Glukoneogeneza je proces stvaranja glukoze iz neugljikohidratnih izvora, a odvija se ponajprije u jetri, manjim dijelom i u bubrezima. Glukoneogeneza zahtijeva nisku razinu inzulina i prisutnost kontraregulatornih hormona (katekolamini, kortizol, hormon rasta) uz odgovarajući dotok supstrata iz mišića i masnoga tkiva. Supstrati iz mišića potrebni za glukoneogenezu su laktati, piruvat te aminokiseline (alanin, glutamin), koji nastaju proteolizom. Glavni supstrati iz masnoga tkiva su glicerol i masne kiseline koje nastaju razgradnjom triglicerida. Same masne kiseline predstavljaju važan izvor energije za stanice u nedostatku glukoze s iznimkom neurona. Fiziološke koncentracije glukoze u plazmi posljedica su koordinacije egzogenoga unosa glukoze, endogene proizvodnje glukoze te periferne utilizacije.
Glavni regulatorni čimbenik održavanja normoglikemije je inzulin, a smanjeno lučenje inzulina prva je obrana od hipoglikemije. Kako nakon obroka dolazi do postupnoga pada vrijedosti glukoze u plazmi, tako dolazi i do smanjenja lučenja inzulina iz β-stanica Langerhansovih otočića. Smanjena razina inzulina smanjuje perifernu utilizaciju glukoze, povećava endogenu proizvodnju glukoze (hepatalna i renalna glukoneogeneza) te osigurava supstrate potrebne za glukoneogenezu potičući lipolizu i proteolizu.
Kada se usprkos smanjenom lučenju inzulina, koncentracija glukoze snizi ispod fiziološke granice, dolazi do pojačanoga lučenja kontraregulatornih hormona, a to su glukagon, adrenalin, kortizol i hormon rasta. Primarni kontraregulatorni hormon je glukagon kojega luče α-stanice Langerhansovih otočića, a glavni mu je mehanizam stimulacija glikogenolize, tj. razgradnje glikogena (skladišni oblik glukoze u jetri) i otpuštanje glukoze u cirkulaciju. Glukagon predstavlja drugu liniju obrane od hipoglikemije. Kada i uloga glukagona postaje nedostatna, počinju se pojačano lučiti katekolamini iz srži nadbubrežne žlijezde, poglavito adrenalin. On stimulira jetrenu i bubrežnu glukoneogenezu i predstavlja treću liniju obrane od hipoglikemije.
Ako stanje hipoglikemije potraje unatoč ovim regulacijskim čimbenicima (dulje od 4 sata), dolazi i do pojačanoga lučenja kortizola i hormona rasta koji smanjuju perifernu utilizaciju glukoze te potiču glukoneogenezu. Oni nemaju tako jak utjecaj kao prethodno opisani čimbenici (učinak čini oko 20 % učinka adrenalina) te ne predstavljaju obrambeni mehanizam akutne hipoglikemije, nego se aktiviraju kasnije.
Osim navedenih mehanizama, bitan je mehanizam i osjećaj gladi koji osobu potiče na egzogeni unos ugljikohidrata. Glikemijski prag kod kojega osobe osjećaju glad i potrebu za unosom hrane obično je između 2,8 i 3,0 mmol/L, iako postoje znatne individualne varijacije. Takav su primjer i bolesnici sa slabo reguliranom šećernom bolesti kod kojih je taj glikemijski prag pomaknut naviše, pa osjećaju simptome hipoglikemije, iako je njihova razina glukoze u plazmi tada u fiziološkim granicama (pseudohipoglikemija) ili pomak glikemijskoga praga naniže kod osoba s učestalim hipoglikemijama te će kod njih izostati osjećaj gladi i simptoma hipoglikemije, iako su vrijednosti glukoze u plazmi ispod fizioloških.
Etiopatogeneza. Hipoglikemija je najčešće povezana s liječenjem šećerne bolesti (nuspojava primjene inzulina i/ili oralnih antihiperglikemika), ali se može javiti i neovisno o njoj (lijekovi, teške bolesti i stanja, deficit kontraregulatornih hormona, endogeni hiperinzulinizam).
Hipoglikemija kod bolesnika sa šećernom bolesti. Hipoglikemija je glavni ograničavajući čimbenik u postizanju dobre kontrole i liječenja bolesnika sa šećernom bolesti: 1) hipoglikemije uzrokuju značajniji morbiditet kod bolesnika sa šećernom bolesti tip 1 i uznapredovale tip 2; 2) hipoglikemije onemogućavaju postizanje zadovoljavajuće glikemije, nužne za sprečavanje razvoja mikrovaskularnih komplikacija; 3) hipoglikemije uvjetuju nastanak tzv. autonomnoga oštećenja uzrokovanoga hipoglikemijom, sidroma koji se očituje zatajenjem uloge zaštitnih mehanizama od hipoglikemije. Hipoglikemije značajno utječu na kvalitetu života osoba sa šećernom bolesti. Bolesnici sa šećernom bolesti tip 1 skloniji su razvoju hipoglikemije od bolesnika sa šećernom bolesti tip 2, no s obzirom na daleko veći broj osoba sa šećernom bolesti tip 2, ukupna incidencija hipoglikemija je kod njih veća. Procjenjuje se da 6 do 10 % bolesnika sa šećernom bolesti tip 1 umre od posljedica hipoglikemija. Razlog zbog kojega su bolesnici sa tipom 1 skloniji hipoglikemijama leži u činjenici da se kod njih radi o apsolutnom manjku inzulina što zahtijeva kompleksnije liječenje inzulinom (više doze, češća primjena). Kod bolesnika sa šećernom bolesti tip 2 hipoglikemija je najčešće rezultat liječenja inzulinom, sulfonilurejom ili glinidima. Hipoglikemija kod takvih bolesnika može biti posljedica više čimbenika: 1) relativni ili apsolutni višak inzulina; 2) poremećaj kontraregulatornih mehanizama; 3) neprepoznavanje simptoma hipoglikemije te 4) razvoj hipoglikemijom uzrokovanoga autonomnog oštećenja (HAAF, engl. hypoglycemia-associated autonomic failure).
Relativni ili apsolutni višak inzulina. Glavni rizik od pojave hipoglikemije predstavlja nastanak relativnoga ili apsolutnoga viška inzulina što može biti povezano s više čimbenika: 1) prevelika i/ili neprimijenjena doza inzulina ili lijekova koji potiču sekreciju inzulina; 2) smanjen ili neodgovarajući unos ugljikohidrata nakon primjene inzulina ili lijekova koji potiču sekreciju inzulina; 3) povećana periferna utilizacija glukoze neovisna o inzulinu (npr. prekomjerna tjelesna aktivnost); 4) povećana osjetljivost na inzulin (nakon poboljšanja glikemije, tijekom noći, nakon tjelesne aktivnosti); 5) smanjena endogena proizvodnja glukoze (nakon unosa alkohola); 6) smanjeni klirens inzulina (npr. bubrežno zatajanje).
Poremećaj kontraregulatornih mehanizama drugi je bitan čimbenik. U uvjetima apsolutnoga deficita endogenoga inzulina izostaje fiziološko smanjenje lučenja inzulina zajedno sa smanjenjem glukoze u plazmi, s obzirom na to da inzulina uopće nema, pa je tako prva linija obrane od hipoglikemije izgubljena. S obzirom na to da je postupno sniženje koncentracije inzulina u plazmi normalan signal za lučenje glukagona, koncentracija glukagona se ne povećava zajedno sa snižavanjem koncentracije glukoze u plazmi, pa je tako i druga linija obrane od hipoglikemije izgubljena. Iako lučenje katekolamina ostaje kao treća linija obrane od hipoglikemije, njihov učinak je atenuiran. Učestale hipoglikemije dovode i do sniženja glikemijskoga praga za aktivaciju pojedinih kontraregulacijskih mehanizama, što stvara veći rizik od nastanka ijatrogene hipoglikemije tijekom agresivnije kontrole glikemije.
Neprepoznavanje simptoma hipoglikemije (nesvjesna hipoglikemija) posljedica je atenuiranoga simpatoadrenalnog odgovora na smanjenje glukoze u plazmi (treća linija obrane od hipoglikemije). To je povezano i sa smanjenim glikemijskim pragom koji se susreće kod opetovanih hipoglikemija. S obzirom na to da se kod ovih bolesnika ne razvijaju simptomi hipoglikemije, osobe ne posežu za povećanim unosom hrane (ugljikohidrata). I takvi bolesnici imaju šesterostruko veći rizik od razvoja ijatrogene hipoglikemije tijekom agresivne glikemijske kontrole u liječenju šećerne bolesti.
Autonomno oštećenje uzrokovano hipoglikemijom (HAAF) posljedica je atenuiranoga simpatoadrenalnog odgovora na hipoglikemiju što dovodi do: 1) smanjenoga lučenja adrenalina i posljedičnoga zakazivanja kontraregulatornih mehanizama te 2) smanjenoga simpatičkoga neuralnog odgovora što se očituje neprepoznavanjem simptoma hipoglikemije. Sve to vodi u hipoglikemiju i tako čini začarani krug.
Hipoglikemija kod bolesnika bez šećerne bolesti. Iako je hipoglikemija najčešće povezana s liječenjem šećerne bolesti (primjena inzulina i lijekova koji potiču lučenje inzulina), može se javiti i u drugim bolestima i stanjima. Prema učestalosti, najčešći uzroci hipoglikemije kod bolesnika bez šećerne bolesti su: 1) lijekovi i alkohol, 2) teške bolesti i stanja, 3) deficit kontraregulatornih hormona i 4) tumori ne-β-stanica, 5) endogeni hiperinzulinizam.
Lijekovi i alkohol. Osim inzulina i lijekova koji potiču lučenje inzulina, i neki drugi lijekovi mogu u rijetkim slučajevima uzrokovati hipoglikemiju. To su ACE inhibitori, blokatori angiotenzinskih receptora, beta blokatori, indometacin, kinoloni i sulfonamidi. Alkohol (etanol) također može uzrokovati hipoglikemiju suprimirajući glukoneogenezu, ali ne i glikogenolizu. Iz toga razloga etanol neće dovesti do hipoglikemije odmah, nego u stanjima kada osoba uzima veće količine alkohola kroz dulje vrijeme uz neodgovarajući unos hrane (ugljikohidrata). U takvim situacijama izostaje odgovarajući egzogeni unos glukoze, troši se glikogen, a glukoneogeneza je suprimirana te se konačno razvija hipoglikemija.
Teške bolesti i stanja. Hipoglikemija se može razviti u sklopu ozbiljnih bolesti i stanja kao što su jetrena i bubrežna insuficijencija, srčano popuštanje, sepsa te kaheksija. S obzirom na to da je jetra dominantan organ u kojemu se vrši endogena proizvodnja glukoze, ali i razgradnja glikogena, brzo progresivna i teška oštećenja jetre (npr. toksični hepatitis) dovode do razvoja hipoglikemije. S obzirom na to da su u endogenu proizvodnju uključeni i bubrezi, zatajenje njihove funkcije može biti povezano s povećanom opasnosti od nastanka hipoglikemije. Ostali razlozi uključuju i smanjen klirens inzulina te smanjenu mobilizaciju supstrata za glukoneogenezu kod bubrežnoga zatajenja. Povezanost srčanoga popuštanja i hipoglikemije nije razjašnjeno, ali se dijelom povezuje s kongestijom jetre i hipoksijom, osobito kod desnostranoga srčanog popuštanja. Sepsa je relativno čest uzrok razvoja hipoglikemija, a posljedica je citokinima inducirane oslabljene glukoneogeneze, ali i povećane periferne utilizacije glukoze. Uz to, hipoperfuzija jetre i bubrega koja se javlja u slučajevima septičkoga šoka dodatno narušava endogenu proizvodnju glukoze. U slučaju gladovanja ili razvoja kaheksija s drugim uzrocima (npr. tumorska kaheksija) dolazi do gubitka zaliha glikogena te iscrpljivanja supstrata za glukoneogenezu, pa se i kod takvih bolesnika u konačnici razvija hipoglikemija.
Deficit kontraregulatornih hormona. Iako ni kortizol ni hormon rasta ne sudjeluju kao obrambeni mehanizmi u razvoju akutne hipoglikemije, njihov deficit (hipopituitarizam, adrenokrotikalna insuficijencija) može biti povezan s razvojem hipoglikemije u slučaju produljenoga gladovanja. Deficit kortizola povezan je s oštećenom glukoneogenezom uslijed smanjene dostupnosti supstrata (tzv. glukoneogeneza limitirana supstratom) te u će se kombinaciji sa smanjenjem glikogena razviti hipoglikemija. Deficit hormona rasta obično je povezan s nastankom hipoglikemije kod manje djece. Produljeno gladovanje, pojačana periferna utilizacija (vježbanje, trudnoća) te smanjena produkcija glukoze (nakon većega unosa alkohola) mogu uzrokovati hipoglikemiju kod odraslih s ranije neprepoznatim hipopituitarizmom. Hipoglikemija nije klinička značajka smanjene produkcije katekolamina nakon bilitaretalne adrenalektomije ako je glukokortikoidna suspstitucija zadovoljavajuća, kao ni u stanjima farmakološke adrenergičke blokade ako su ostali glukoregulatorni sistemi uredni.
Hipoglikemija i tumori ne-β-stanica. Veliki mezenhimalni i epitelni tumori (adenokarcinom, karcionid, hepatoadenom) mogu uzrokovati hipoglikemiju stvarajući tvari slične inzulinu, kao što je inzulinu sličan faktor rasta II (IGF-II). Njihov učinak jednak je učinku inzulina, dok je sama sekrecija inzulina suprimirana. Kirurška resekcija tumora može smanjiti učestalost hipoglikemija, kao i liječenje glukortikoidima i/ili hormonom rasta.
Endogeni hiperinzulinizam. Pojačana endogena produkcija inzulina može biti posljedica: 1) tumora ili hiperplazije β-stanica Langerhansovih otočića; 2) stvaranja antitijela na inzulin ili inzulinske receptore; 3) primjene lijekova koji potiču proizvodnju inzulina u β-stanicama (npr. sulfonilureja) te 4) ektopično endogeno stvaranje inzulina. Temeljna patofiziološka značajka endogene hiperinzulinemije uzrokovane tumorom/hiperplazijom β-stanica ili primjenom lijekova koji potiču lučenje inzulina je nemogućnost supresije inzulinske sekrecije i, što se može dokazati mjerenjem vrijednosti inzulina, C-peptida te proinzulina za vrijeme hipoglikemije. Kritične, dijagnostički značajne vrijednosti kod hipoglikemije uzrokovane hiperinzulinizom su vrijednosti inzulina ≥ 3 μU/mL (≥ 18 pmol/L), C-peptida ≥ 0.6 ng/mL (≥ 0.2 nmol/L) te proinzulina ≥ 5.0 pmol/L pri vrijednostima glukoze u plazmi manjima od 3,0 mmol/L uz prisutnost simptoma hipoglikemije.
Ostali, rjeđi uzroci hipoglikemija su stanja nakon želučanih operacija, hiperplazija β-stanica, autoimuna hipoglikemija te hipoglikemija uzrokovana antitijelima na inzulinske receptore. Ukupno gledano, radi se o izuzetno rijetkim uzrocima hipoglikemija. Hipoglikemija nakon želučanih operacija (gastrektomija, vagotomija, piloroplastika, Billroth II) očituje se u sklopu tzv. kasnog dumping sindroma. Javlja se 1 do 3 sata nakon obilnijega ugljikohidratnog obroka nakon brze propulzije sadržaja u proksimalni dio tankoga crijeva nakon čega slijedi ubrzana i povećana apsorpcija glukoze u krv što potiče pojačano lučenje inzulina i nastanak hiperinzulinemije. Smatra se da u tom hiperinzulinskom odgovoru ulogu imaju gastrointestinalni hormoni kao što je GLP-1. Kod manjega se broja bolesnika s organskom hiperinzulinemijom ne može dokazati tumor β-stanica (inzulinom), ali se patohistološki otkriju hipertrofija i hiperplazija β-stanica Langerhansovih otočića, i to se naziva neinzulinomski pankreasni hipoglikemični sindrom. Među rjeđe oblike hipoglikemije ubrajamo i autoimunu hipoglikemiju povezanu s prisustvom antitijela (obično IgG klase) koja mogu vezati inzulin te ga kasnije postupno otpuštati. To se često susreće pri liječenju metimazolom, ali i u sklopu nekih autoimunih bolesti kao što su reumatoidni artritis, sistemski lupus, polimiozitis i sl. Hipoglikemiju mogu uzrokovati i antitijela na inzulinske receptore koja se mogu vezati na njih i djelovati agonistički.
Postprandijalna hipoglikemija i hipoglikemija natašte. Ovisno o vremenu kada se hipoglikemija javlja, razlikujemo postaprandijalnu (reaktivnu) hipoglikemiju te hipoglikemiju natašte. Postprandijalna hipoglikemija javlja se nekoliko sati nakon obroka, dok se hipoglikemija natašte javlja u vrijeme gladovanja. To razlikovanje može pomoći pri diferencijalnoj dijagnozi hipoglikemija. Postprandijalna hipoglikemija najčešće je posljedica želučanih operacija (gastrektomija, resekcija želuca, vagotomija) kada dolazi do nagloga pražnjenja želuca te brze apsorpcije glukoze i prekomjernoga lučenja izulina, nakon čega slijedi brži pad koncentracije glukoze od koncentracije inzulina. Ostali uzroci obično su povezani s dječjom dobi: nasljedna netolerancija na glukozu, galaktozemija te nasljedna osjetljivost na leucin. Hipoglikemija natašte posljedica je neravnoteže između proizvodnje glukoze i periferne utilizacije glukoze (tumori β-stanica, ekstrapankreatični tumori i dr.).
Klinička slika. Hipoglikemija se očituje neuroglikopenijskim i autonomnim (neurogenim) simptomima i znakovima. Neuroglikopenične manifestacije posljedica su smanjenoga dotoka glukoze neuronima i njihovoga posljedičnoga poremećenoga metabolizma i funkcioniranja. Ponajprije uključuju promjene u ponašanju, konfuziju, narušenu koncentraciju, glavobolju, vrtoglavicu, zamagljen vid i slabljenje motoričkih funkcija. Autonomne manifestacije posljedica su simpatoadrenergičkoga odgovora i pojačanoga lučenja adrenalina. Podrazumijevaju znojenje, tremor, tahikardiju, nemir i glad. Naposljetku dolazi do postupnoga jačanja poremećaja svijesti sve do stanja kome, a ako se dalje nastavi i produbi hipoglikemija, moguć je i smrtni ishod. Kada se hipoglikemija razvija, postupno dominiraju simptomi od strane središnjega živčanog sustava, a adrenergički simptomi mogu proći nezapaženo, a kod bržega razvoja hipoglikemije, adrenergički simptomi mogu biti jače izraženi. Također, kod bolesnika sa šećernom bolesti s razvijenom dijabetičkom neuropatijom, adrenergički simptomi mogu izostati. Koncentracije glukoze u plazmi kod kojih hipoglikemija postaje manifestna nije kod svih ista, a kod osoba koje ne boluju od šećerne bolesti obično nastaju kada vrijednost glukoze u venskoj krvi padne ispod 2,5 mmol/L.
Dijagnostički pristup. Dijagnostička obrada hipoglikemije podrazumijeva: 1) određivanje glukoze u plazmi; 2) test 72-satnoga gladovaja; 3) petosatni oralni test tolerancije glukoze; 4) test supresije C-peptida; 5) određivanje inzulinskih antitijela i 6) određivanje glikiranoga hemoglobina.
Određivanje glukoze u plazmi. U dijagnostici same hipoglikemije potrebno je dokumentirati nisku koncentraciju glukoze u plazmi za vrijeme simptoma koji odgovaraju hipoglikemiji, kao i njihovo povlačenje nakon unosa ugljikohidrata. To je najpouzdaniji parametar potvrde da se radi o hipoglikemiji. Samo mjerenje niskih koncentracija glukoze u plazmi, osobito kod zdravih asimptomatskih pojedinaca ne mora ništa značiti s obzirom na velike individualne razlike. Normalne vrijednosti glukoze u plazmi za vrijeme pojave simptoma koji bi mogli odgovarati hipoglikemiji isključuju njezino postojanje i uzrok treba tražiti dalje. Ako je Whippleov trijas pozitivan, potrebno je provesti daljnju dijagnostičku obradu.
Test 72-satnoga gladovanja. S obzirom na to da se često ne može postići mjerenje glukoze u plazmi (ali i drugih parametara) u vrijeme kada se jave simptomi hipoglikemije, a postoji pozitivna anamneza na njih, potrebno je provesti test kojime se bolesnika svjesno nastoji uvesti u hipoglikemiju, kako bi se to laboratorijski dokumentiralo. S tom se svrhom provodi test 72-satnoga gladovanja. Taj se test provodi i kod svih bolesnika s pozitivnim Whippleovim trijasom. Test se provodi u kontroliranim uvjetima prema standardiziranom protokolu, a podrazumijeva podvrgavanje bolesnika 72-satnom gladovanju u kontroliranim uvjetima. Protokol uključuje sljedeće: 1) s početkom testa potrebno je prestati unosti hranu i pića koja sadrže kalorijsku vrijednost, kao i sve lijekove koji utječu na glikemiju; 2) bolesnici mogu konzumirati piće bez kalorija i kofeina; 3) bolesnici moraju biti aktivni tijekom perioda budnosti; 4) potrebno je svakih 6 sati mjeriti vrijednosti glukoze u plazmi, inzulina, C-peptida i β-hidroksibutirata (u istom uzorku venske krvi), sve dok se koncentracija glukoze u plazmi ne snizi do 3,3 mmol/L, a zatim iste parametre mjeriti svakih 1 do 2 sata; 5) test se završava nakon isteka perioda od 72 sata ili ranije, ako se koncentracija glukoze u plazmi spusti ispod 2,5 mmol/l i pacijent pri tome ima odgovarajuću simptomatologiju, ili ispod 3 mmol/L ako je Whippleov trijas ranije dokazan; 6) na kraju testa potrebno je izmjeriti vrijednosti glukoze u plazmi, inzulina, C-peptida, β-hidroksibutirata (β-HBT) te sulfonilureje - SLF (u istom venskom uzorku). Interpretacija 72-satnoga testa gladovanja prikazana je u Tablici 8.11.
|
Tablica 8.11. Primjeri tumačenja nalaza 72-satnog testa gladovanja
|
|
Vrsta hipoglikemije
|
Simptomi i znakovi
|
Glukoza mg/dL
|
Inzulin μU/mL
|
C-peptid pmol/L
|
Proinzulin pmol/L
|
β-HBT mmol/L
|
SLF u plazmi
|
|
Normalni nalaz
|
Ne
|
≥ 40
|
< 3
|
< 200
|
< 5
|
> 2,7
|
Ne
|
|
Inzulinom (tumor)
|
Da
|
≤ 45
|
≥ 3
|
≥ 200
|
≥ 5
|
≤ 2,7
|
Ne
|
|
SLF-inducirana hipoglikemija
|
Da
|
≤ 45
|
≥ 3
|
≥ 200
|
≥ 5
|
≤ 2,7
|
Da
|
|
Hipoglikemija posredovana IGFII
|
Da
|
≤ 45
|
≤ 3
|
< 200
|
< 5
|
≤ 2,7
|
Ne
|
|
Inzulin neovisna hipoglikemija
|
Da
|
≤ 45
|
< 3
|
< 200
|
< 5
|
> 2,7
|
Ne
|
|
Nehipoglikemijski poremećaj
|
Da
|
≥ 40
|
< 3
|
< 200
|
< 5
|
> 2,7
|
Ne
|
Petosatni test tolerancije glukoze. Ovaj test se provodi kod osoba sa sumnjom na postprandijalnu hipoglikemiju, tj. kod bolesnika kod kojih se javljaju simptomi hipoglikemije pet sati od uzimanja obroka. Kod njih se test provodi tako da im se da obrok sličan onome koji dovodi do pojave simptoma te ih se prati kroz sljedećih pet sati. Test je pozitivan ako se kod njih razviju simptomi hipoglikemije te se tada izmjere niske vrijednosti glukoze u plazmi. Sam pozitivan test ne potvrđuje dijagnozu, nego usmjerava na daljnju dijagnostiku (72-satni test gladovanja). Pozitivan petosatni test tolerancije glukoze i negativan 72-satni test gladovanja mogu imati neki pacijenti s inzulinomom i hiperplazijom β-stanica.
Test supresije C-peptida. Ovaj se test temelji na činjenici da je sekrecija iz β-stanica (mjerena vrijedostima C-peptida) jače suprimirana tijekom hipoglikemije kod pacijenata s inzulinomom nego kod zdravih osoba. Test se rijetko izvodi, a interpretacija ovisi o indeksu tjelesne mase, kao i dobi pacijenta.
Određivanje inzulinskih antitijela. Detekcija takvih antitijela opravdana je kod bolesnika koji su u terapiji koristili animalne inzuline, s obzirom na to da humani inzulini nisu toliko antigeni da bi tijelo produciralo antitijela na njih. Određivanje ovih antitijela opravdano je i pri sumnji na autoimunu hipoglikemiju.
Određivanje glikiranoga hemoglobina. Glikirani hemoglobin značajno je niži kod bolesnika s inzulinomom nego kod zdravih pojedinaca što može pomoći u diferencijalnoj dijagnozi hipoglikemije.
Pristup bolesniku s hipoglikemijom. Ako se radi o hipoglikemiji povezanoj sa šećernom bolesti, obično nije potrebna daljnja dijagnostička obrada jer se ona smatra posljedicom liječenja (primjena inzulina ili lijekova koji potiču lučenje inzulina). Tek ako se i nakon revizije i optimizacije terapije hipoglikemije i dalje javljaju, potrebno je provesti opsežniju dijagnostičku obradu. Kod bolesnika s hipoglikemijom koja nije vezana uz šećernu bolest potrebno je također provesti detaljniju dijagnostičku obradu. Potrebno je uzeti detaljnu anamnezu i napraviti klinički pregled te pristupiti ranije opisanim dijagnostičkim testovima za hipoglikemiju. U prvom redu je kod tih pacijenata potrebno isključiti hipoglikemiju kao posljedicu uzimanja lijekova, alkohola ili težih bolesti i stanja. Ako su ti uzroci isključeni, potrebno je provesti probir na bolesti hipofize i nadbubrežne žllijezde koje mogu dovesti do deficita kontraregulatornih hormona, što može uzrokovati hipoglikemiju. Ako se postavi sumnja na inzulinom, potrebno je napraviti odgovarajuću dijagnostičku evaluaciju u tom smjeru (ultrazvuk i kompjuterizirana tomografija trbuha, oktreoscan).
Liječenje. Terapijski pristup bolesniku u hipoglikemiji temelji se na akutnom ispravljanju nastale hipoglikemije te prevenciji nastanka nove epizode hipoglikemije. Kada se otkrije uzrok hipoglikemije, potrebno je pristupiti etiološkom, specifičnom liječenju.
Akutno liječenje hipoglikemije. Ako se radi o suradljivom bolesniku koji može enteralno unositi hranu, preporuča se peroralni unos glukoze (preporučena početna doza je 20 g glukoze u obliku slatkiša, slatke tekućine i sl.). Ako bolesnik nije suradljiv, glukoza mu se ordinira parenteralno. To najčešće podrazumijeva bolusnu primjenu 50 %-tne glukoze po 10 ml intravenski uz praćenje glikemije do nastanka euglikemije te potom nastavak infuzije 5 %-tne ili 10 %-tne glukoze zbog održavanja euglikemije. Druga je opcija primjena glukagona u dozi od 1 mg intramuskularno ili supkutano. S obzirom na to da glukagon postiže euglikemiju razgradnjom glikogena, u stanjima kod kojih je zaliha glikogena iscrpljena (dugotrajno gladovanje, alkohol) njegova primjena neće biti učinkovita. Kod bolesnika kod kojih je hipoglikemija uzrokovana lijekovima koji potiču lučenje inzulina korisna može biti primjena somatostatinskog analoga oktreotida jer će dovesti do smanjenoga lučenja inzulina. Taj učinak je kratkotrajan pa bolesnici čim se oporave trebaju uzimati obroke bogate ugljikohidratima (ili primijeniti infuzije glukoze) kako bi se napunile zalihe glikogena.
Prevencija hipoglikemije. Prevencija ponovnoga nastupa hipoglikemije zahtijeva poznavanje mehanizma nastanka hipoglikemije. Kod hipoglikemija povezanih s primjenom lijekova potrebna je reevaluacija doze lijeka ili je potrebno ukinuti davanje lijeka. Hipoglikemija uzrokovana sulfonilurejom može trajati do nekoliko dana pa je potreban poseban nadzor nad bolesnicima (obično u bolničkim uvjetima) te stalna nadoknada glukoze. Ostali uzorci hipoglikemija zahtijevaju specifične terapije: nadoknada kortizola i/ili hormona rasta kod hipoptuitarizma ili adrenokrotikalne insuficijencije; kirurško-onkološki tretman ne-β-tumora povezanih s hipoglikemijom; operativno liječenje inzulinoma; primjena kortikosteroida kod autoimune hipoglikemije i sl.
POREMEĆAJI METABOLIZMA LIPIDA (HIPERLIPIDEMIJE)
Poremećaji metabolizma lipida, odnosno hiperlipidemije, među najčešćim su metaboličkim poremećajima koje susrećemo u svakodnevnoj kliničkoj praksi, čija važnost proizlazi iz jake povezanosti s procesom aterogeneze i nastankom kardiovaskularnih oboljenja. U daljnjem tekstu bit će prikazan metabolizam egzogenih i endogenih lipida, najčešće primarne hiperlipidemije, uzroci sekundarnih hiperlipidemija kao i klinički pristup bolesniku s hiperlipidemijom koji obuhvaća kliničku prezentaciju, dijagnostički postupak i liječenje.
METABOLIZAM LIPIDA I HIPERLIPIDEMIJA
Lipidi (masti) imaju različite uloge u organizmu: oni čine gradivne stanične elemente, sudjeluju u sintezi različitih biološki aktivnih molekula (hormoni) te osiguravaju energiju. U organizmu dolaze u tri glavna oblika: kolesterol, trigliceridi i fosfolipidi, a mogu biti unešeni hranom ili sintetizirani u organizmu iz drugih, nelipidnih molekula. S obzirom na to da su lipidi netopljive i hidrofobne molekule, u tjelesnim se tekućinama prenose putem proteinskih kompleksa koji se nazivaju lipoproteini. Oni se sastoje od dva osnovna dijela: lipidni (središnji) i proteinski (površinski) dio. Proteini koji ulaze u sastav lipoproteina nazivaju se apoproteinima (Apo) te, osim što osiguravaju odgovarajuću strukturu lipoproteina, imaju važnu ulogu kao kofaktori pojedinih enzima koji sudjeluju u samom metabolizmu (transportu) lipida. Ovisno o gustoći lipoproteini se dijele u pet skupina: hilomikroni, lipoproteini vrlo male gustoće (VLDL, od engl. very low density lipoprotein), lipoproteini srednje gustoće (IDL, od engl. intermediate density lipoprotein), lipoproteini male gustoće (LDL, od eng. low density lipoprotein) te liporoteini velike gustoće (HDL, engl. high density lipoprotein). Njihove osnovne značajke prikazane su u Tablici 8.12.
|
Tablica 8.12. Karakteristike lipoproteina
|
|
Vrsta lipoproteina
|
Sastav lipoproteina: lipidi
|
Sastav lipoproteina: apoproteini
|
Dijametar (nm)
|
Gustoća (g/ml)
|
|
Hilomikroni
|
Trigliceridi iz crijeva
|
Apo B48, Apo CI, CII, CIII, Apo E
|
80 – 500
|
< 1.006
|
|
VLDL
|
Trigliceridi iz jetre
|
Apo B100, Apo CII, CIII, Apo E
|
30 – 80
|
< 1.006
|
|
IDL
|
Kolesterol, trigliceridi
|
Apo B100, Apo E
|
25 – 35
|
1.006 - 1.019
|
|
LDL
|
Kolesterol
|
Apo B100
|
18 – 25
|
1.019 - 1.063
|
|
HDL
|
Kolesterol, fosfolipidi
|
Apo AI, AII, Apo CI, CII, CIII
|
5 – 12
|
1.063 - 1.210
|
Metabolizam egzogenih lipida. Lipidi koji se u organizam unesu hranom u crijevu su razgrađeni na kolesterol i slobodne masne kiseline koji se potom unose u enterocite u kojima se vežu na Apo B48 te tvore hilomikrone koji dospijevaju u cirkulaciju. U cirkulaciji hilomikroni primaju i druge bitne apoproteine kao što su Apo C II i drugi. Hilomikroni dospijevaju u periferna tkiva, osobito u masno i mišićno tkivo te se tamo uz pomoć enzima lipoprotein lipaze koja se nalazi vezana za endotel kapilara te hormonski ovisne lipaze koja se nalazi u masnom tkivu razgrađuju na slobodne masne kiseline koje se potom dalje skladište (masno tkivo) ili koriste za energiju (mišićno tkivo). Za aktivaciju lipoprotein lipaze nužan je Apo CII koji se nalazi na površini hilomikrona. Ostatci hilomikrona koji poglavito sadrže estere kolesterola uklonjeni su putem jetre uz pomoć Apo E.
Metabolizam endogenih lipida. Osim egzogenoga unosa, lipidi mogu nastati i unutar organizma iz nelipidnih molekula procesom koji se naziva lipogeneza, a odvija se u jetri. Na taj se način višak ugljikohidrata slijedom metaboličkih reakcija pretvara u trigliceride koji se potom putem lipoproteina prenose do perifernih organa (mišićno i masno tkivo). Osim triglicerida, u jetri (ali i u drugim tkivima) moguća je i endogena sinteza kolesterola pri čemu najvažniju ulogu ima enzim HMG-CoA reduktaza. Sintetizirani trigliceridi u jetri vežu se na Apo B100 što omogućuje mikrosomalni prijenosi protein (MTP) te tako nastaju VLDL čestice, a koje u svom sastavu sadrže i druge apoproteine koji imaju specifične učinke. Osim dominantnih triglicerida, VLDL čestice sadrže i kolesterol. U kapilarama perifernih tkiva lipoprotein lipaza koju aktivira Apo CII koja se nalazi u sastavu VLDL-a dovodi do njegove razgradnje pri čemu se oslobađaju slobodne masne kiseline, a VLDL se pretvara u IDL koji sada sadrži manje količine triglicerida i kolesterol. IDL se dalje odstranjuje na dva načina: 1) ulaskom u hepatocite posredovano LDL receptorima na površini hepatocita koji prepoznaju Apo B100 i Apo E (neki ih autori zbog toga nazivaju i B/E receptorima) ili 2) razgrađuje ih hepatalna lipaza koja se također nalazi na površini hepatocita pri čemu od IDL-a nastaje LDL (uklanja se ostatak triglicerida i apoproteina osim Apo B100). Prema tome, nastali LDL sastoji se samo od kolesterola i Apo 100. Najveći dio LDL čestica biva prepoznat od strane LDL receptora i unesen u hepatocite gdje se dalje razgrađuje na sastavne dijelove, dok se manji dio uklanja putem "stanica čistača" - makrofaga i glatkih mišićnih stanica. LDL receptore posjeduju i drugi organi kao što su npr. nadbubrežne žlijezde i gonade gdje LDL čestice osiguravaju kolesterol potreban za sintezu hormona. Uz LDL receptore opisuje se i PCSK9 enzim (engl. proprotein convertase subtilisin-like/kexin typ 9) koji je odgovoran za degradaciju LDL receptora te njegova prekomjerna ekspresija rezultira smanjenjem broja LDL receptora na površini stanica što je povezano s nastankom hiperlipidemija. Osim toga, LDL čestice ulaze i u subendotelni prostor čime se objašnjava njihov izrazit aterogeni potencijal („loš kolesterol“). Osim navedenih lipoproteina, u cirkulaciji nalazimo još jedan oblik - HDL. On nastaje u hepatocitima i crijevima, sadrži fosfolipide te dominantno Apo E te Apo A te ima glavnu karakteristiku da vrlo brzo izmjenjuje lipide i apoproteine s drugim lipoproteinima. Na taj način HDL donira Apo CII i druge bitne apoproteine, a akceptira kolesterol i trigliceride koji dolaze iz membrana stanica ili s površine drugih lipoproteina. HDL se uklanja putem jetre uz pomoć receptora čistača za HDL (SR-B1). Na taj način HDL kupi višak kolesterol s periferije i prenosi ga do jetre čime ostvaruje svoj pozitivan efekt u procesu aterogeneze („dobri kolesterol“). Osim toga, kolesterol iz HDL-a se može ukloniti i njegovom izmjenom s trigliceridima iz VLDL čestice pri čemu se VLDL pretvara u IDL, a dijelom bude razgrađen u jetri.
Hiperlipidemija (hiperlipoproteinemija) je naziv koji opisuje stanje povišenih vrijednosti lipida u krvi, a prema vrsti povišenih lipida dijeli se na hiperkolesterolemiju, hipertrigliceridemiju ili miješanu hiperlipidemiju. Ovisno o uzroku, dijele se na primarne hiperlipidemije koje nastaju uslijed genetskih poremećaja te sekundarne hiperlipidemije koje nastaju kao posljedica određenih bolesti i stanja. Iako se danas rijetko koristi, u Tablici 8.13. je prikazana Fredricksonova klasifikacija hiperlipidemija ovisno o povišenim vrijednostima pojedinih oblika lipoproteina.
|
Tablica 8.13. Fredricksonova klasifikacija hiperlipidemija
|
|
Tip hiperlipidemije
|
Povećana koncentracija
|
|
Lipoproteini
|
Lipidi
|
|
I
|
Hilomikroni
|
Trigliceridi
|
|
IIa
|
LDL
|
Kolesterol
|
|
IIb
|
LDL, VLDL
|
Kolesterol, trigliceridi
|
|
III
|
IDL
|
Trigliceridi, kolesterol
|
|
IV
|
VLDL
|
Trigliceridi
|
|
V
|
VLDL, hilomikroni
|
Trigliceridi, kolesterol
|
PRIMARNE HIPERLIPIDEMIJE
Primarne hiperlipidemije nastaju zbog genetski nasljednih poremećaja, a mogu se očitovati kao izolirana hiperkolesterolemija (obiteljska hiperkolesterolemija, obiteljski manjak Apo B100, poligenska hiperkolesterolemija), izolirana hipertrigliceridemija (obiteljska hipertrigliceridemija, manjak lipoproteinske lipaze, manjak Apo CII) te miješana hiperlipidemija (obiteljska miješana hiperlipidemija, obiteljska disbetalipoproteinemija).
IZOLIRANA HIPERKOLESTEROLEMIJA
Obiteljska hiperkolesterolemija nastaje uslijed mutacije gena za LDL receptor (LDLR gen) koji se nalazi na 19. kromosomu, a do danas je opisano više od 1500 različitih mutacija. Bolest se nasljeđuje autosomno-dominantno: homozigoti imaju potpun nedostatak LDL receptora i bolest se očituje u djetinjstvu, dok heterozigoti imaju smanjen broj ili funkcionalni defekt LDL receptora što se klinički očituje kasnije (slučajni laboratorijski nalaz hiperkolesterolemije, rani kardiovaskularni incident). Učestalost heterozigotne obiteljske hiperkolesterolemije je velika i iznosi 1:250 do 1:500, dok učestalost homozigotne iznosi 1:1000000. Patofiziološki gledano, zbog nemogućnosti ulaska LDL-a u stanicu, on se nakuplja u krvi, dok se u stanicama kompenzatorno pojačava aktivnost HMG-CoA-reduktaze koja povećava endogenu sintezu kolesterola. Posljedično dolazi do ubrzane aterogeneze i razvoja kardiovaskularnih komplikacija. Većina bolesnika s heterozigotnom hiperkolesterolemijom razvije neki od kardiovaskularnih incidenata prije 60. godine života, najčešće akutni infarkt miokarda. Također, u kliničkoj slici nalazimo ksantome i ksantelazme. Dijagnoza se postavlja na temelju laboratorijskih nalaza (povišen LDL kolesterol > 4 mmol/L uz uredne trigliceride i uredan ili snižen HDL kolesterol) te pozitivnu obiteljsku anamnezu (hiperkolesterolemija i/ili rani kardiovaskularni incident prije 55 godine kod muškaraca, odnosno 60 godina kod žena). Genetska analiza je dostupna (DNA analiza prisutnosti funkcionalne mutacije za LDL receptore, apoliprotein ß i PCSK9), ali se za postavljanje dijagnoze koristi validirani skup kriterija koji uključuje obiteljsku anamnezu ranih kardiovaskularnih događaja, osobnu anamnezu kardiovaskularnih događaja, razinu neliječenoga LDL kolesterola te znakove kao što su prisutnost ksantoma i ksantelazmi prije 45. godine života (Dutch Lipid Clinic Network Score (DLCNS)).
Obiteljski manjak Apo B100 je autosomno-dominantan poremećaj koji nastaje uslijed mutacije gena za Apo B100 koji se nalazi na 2. kromosomu pri čemu nastaje strukturno ili funkcionalno promijenjen Apo B100 što rezultira smanjenjem afiniteta LDL čestica prema LDL receptoru pa dolazi do nakupljanja LDL-a u plazmi i nastanka hiperkolesterolemije (konačni ishod je isti kao i kod obiteljske hiperkolesterolemije). Homozigoti imaju više vrijednosti ukupnoga i LDL kolesterola te se bolest očituje ranije, dok heterozigoti imaju niže vrijednosti i bolest se očituje kasnije. Kliničke manifestacije i dijagnostički postupak jednaki su kao i kod obiteljske hiperkolesterolemije, a razlikovanje ova dva poremećaja moguće je samo na osnovi genetske analize što se ne provodi u kliničkoj praksi s obzirom na to da je liječenje istovjetno.
Poligenetska hiperkolesterolemija je oblik nasljedne izolirane hiperkolesterolemije koja fenotipski (klinički) nalikuje prethodno navedenim poremećajima, ali nije uzrokovana mutacijom jednoga (većeg) gena, nego je posljedica više mutacija (manjih) gena kao što su APOE, ABCG5, ABCG8, LIPA. U razvoju ove hiperkolesterolemije bitnu ulogu imaju i vanjski rizični čimbenici (prehrana, tjelesna aktivnost i sl.). Hiperkolesterolemija je obično umjerena, a rizik od kardiovaskularnoga incidenta nešto je niži nego kod bolesnika s obiteljskom hiperkolesterolemijom ili manjkom Apo B100 te je karakterističan izostanak tetivnih ksantoma (diferencijalno-dijagnostički podatak). Za potpunu potvrdu dijagnoze potrebna su genetska ispitivanja (ne rade se rutinski).
IZOLIRANA HIPERTRIGLICERIDEMIJA
Obiteljska hipertrigliceridemija je naziv za nasljedni poremećaj metabolizma triglicerida koji se očituje izoliranom hipertrigliceridemijom uz odsutnost mogućih sekundarnih uzroka. Točan genetski defekt nije poznat, a sve više se govori o mogućoj poligenetskoj etiologiji (višestruke genske mutacije) pa se i predlaže promjena naziva u poligenetsku hipertrigliceridemiju. Vrijednosti LDL kolesterola su uredne ili blago povišene, dok su vrijednosti HDL kolesterola snižene. Dijagnozu podržava nalaz hipertrigliceridemije kod bližih srodnika. Utjecaj na pojavu kardiovaskularnih incidenata nije razjašnjen, no često je udružena s debljinom, šećernom bolešću tip 2 te inzulinskom rezistencijom, dok povećanu incidenciju akutnoga pankreatitisa nalazimo u ranoj životnoj dobi.
Obiteljski manjak lipoproteinske lipaze je autosomno-recesivan poremećaj koji nastaje uslijed mutacije LPL gena koji se nalazi na 8. kromosomu što rezultira smanjenom aktivnošću enzima lipoprotein lipaze i nakupljanjem hilomikrona u cirkulaciji. Procijenjena incidencija iznosi jedan na milijun stanovnika. Bolest klinički karakteriziraju eruptivni ksantomi, hepatosplenomegalija te recidivni pankreatitis. Kliničke manifestacije pojavljuju se već od djetinjstva. S obzirom na to da hilomikroni imaju malen aterogeni potencijal, poremećaj nije povezan s većom učestalošću kardiovaskularnih događaja i akceleriranom aterosklerozom. Dijagnoza se postavlja na temelju kliničke slike i laboratorijskih nalaza, rijetko se primjenjuje test stimulacijom heparinom kojim se dokazuje nedostatna ili smanjena aktivnost lipoprotein lipaze.
Obiteljski manjak Apo CII je autosomno-recesivan poremećaj koji nastaje uslijed mutacije APOC2 gena koji se nalazi na 19. kromosomu što rezultira strukturnim ili funkcionalnim poremećajem apoproteina CII koji ima ključnu ulogu u aktivaciji lipoprotein lipaze i hidrolizi hilomikrona. Posljedično tome, aktivacija lipoprotein lipaze je nezadovoljavajuća te se javljaju patofiziološke i kliničke posljedice kao i kod obiteljskoga manjka lipoproteinske lipaze (nagomilavanje hilomikrona, hipertrigliceridemija, recidivni pankreatitisi). Eruptivni ksantomi nisu karakteristični za ovu bolest. Primjena svježe smrznute plazme smanjuje vrijednosti triglicerida budući da se u njoj nalazi dosta Apo CII.
MIJEŠANA HIPERLIPIDEMIJA
Obiteljska miješana hiperlipidemija označava nasljedni poremećaj metabolizma lipida koji se očituje povišenim vrijednostima triglicerida i kolesterola, a nađe se kod oko 1:100 do 1:200 osoba u općoj populaciji. Točan genetski defekt nije poznat, a smatra se da se radi o poligenetskoj etiologiji. Radi se o značajnom rizičnom čimbeniku u razvoju kardiovaskularnih komplikacija ateroskleroze. Dijagnoza se postavlja na temelju laboratorijskih nalaza kombinirane hipertrigliceridemije i hiperkolesterolemije uz pozitivan anamnestički podatak o hiperlipidemiji i/ili ranom kardiovaskularnom incidentu kod bližih srodnika.
Obiteljska disbetalipoproteinemija je autosomno-dominantan poremećaj u kojemu nalazimo genetski defekt APOE gena (homozigoti E2/E2) zbog čega nastaje strukturno i/ili funkcionalno izmijenjen apoprotein E koji je nužan za vezanje IDL-a, LDL-a i hilomikrona za receptore na hepatocitima. Posljedično dolazi do nastanka miješane hiperlipidemije. Klinički nalaz uključuje ksantome, rani kardiovaskularni događaj, palmarne strije žućkaste boje te tuberoeruptivne ksantome.
SEKUNDARNE HIPERLIPIDEMIJE
Sekundarne hiperlipidemije su stečene hiperlipidemije koje se javljaju uz neke bolesti i stanja ili se javljaju uslijed uzimanja nekih lijekova (prikazano u Tablici 8.14.). Mehanizmi kojima navedeni uzroci dovode do hiperlipidemije opisani su u odgovarajućim dijelovima udžebnika.
|
Tablica 8.14. Najčešći uzroci sekundarne hiperlipidemije
|
|
Bolesti i stanja
|
Lijekovi
|
|
Šećerna bolest
Pretilost
Hipotireoza
Alkoholizam
Nefrotski sindrom
Bolesti jetre (kolestaza)
Trudnoća
Imunološke bolesti (SLE)
|
Diuretici
Beta blokatori
Anabolni steroidi
Estrogeni
Glukokortikoidi
Klasični antipsihotici
Retinoidi
Antivirotici (inhibitori proteaza)
|
KLINIČKI PRISTUP BOLESNIKU S HIPERLIPIDEMIJOM
Klinička slika. U svojoj osnovi hiperlipidemija predstavlja asimptomatsku bolest koja je povezana s različitim komplikacijama (kardiovaskularna bolest, pankreatitis i sl.). Kod nekih se bolesnika mogu naći karakteristični znakovi hiperlipidemije: ksantomi, ksantelazmi, prsten rožnice (arcus corneae) te lipemia retinalis.
Ksantomi nastaju kao posljedica nakupljanja lipida u potkožnom tkivu, a očituju se kao žućkasto-smećkasti plakovi koji se izdižu iznad razine kože. Razlikujemo tuberozne ksantome koji se obično nalaze na mjestima izloženim traumama (tetive, laktovi, koljena, zglobovi), a očituju se kao različito veliki plakovi žute boje te eruptivne ksantome koji nastaju naglo, tamno žute do narančaste boje koji su okruženi eritemom uslijed lokalne upalne reakcije, a distribuiraju se po trupu i ekstenzornim stranama udova.
Ksantelazme predstavljaju jasno ograničene, uzdignute bjeličaste ili žućkaste ploče koje se pojavljuju u periorbitalnoj regiji (najčešće na vjeđama), a nastaju uslijed odlaganja lipida.
Prsten rožnice (arcus corneae) nastaje kao posljedica odlaganja lipida u rožnicu stvarajući karakteristični bjeličasti prsten uz rub šarenice.
Lipemia retinalis označava karakteristične promjene na krvnim žilama mrežnice koje se mogu vidjeti uslijed hipertrigliceridemije. Zbog povećane koncentracije triglicerida u krvnim žilama one na pregledu daju karakteristično bjeličasto prosijavanje.
Dijagnoza hiperlipidemija temelji se na anamnezi, fizikalnom pregledu te laboratorijskoj dijagnostici. Klasični lipidogram podrazumijeva određivanje vrijednosti triglicerida, ukupnoga kolesterola te LDL i HDL kolesterola, dok prošireni lipidogram uključuje i određivanje ne-HDL kolesterola te apoproteina A i B. Genetska analiza pri sumnji na primarnu hiperlipidemiju nema veći značaj u svakodnevnoj kliničkoj praksi.
Anamneza i fizikalni pregled igraju važnu ulogu u postavljanju dijagnoze hiperlipidemije, osobito u razlučivanju primarne i sekundarne etiologije: podaci o ranijim bolestima i stanjima, funkcijama i navikama, uzimanju lijekova, postojanju hiperlipidemije i/ili pojave ranoga kardiovaskularnog incidenta kod bližih rođaka i sl. Prilikom kliničkoga pregleda mogu se uočiti klinički znakovi koji upućuju na hiperlipidemiju (pretilost, ksantomi, ksantelazmi i sl.).
Laboratorijsko mjerenje lipida u krvi (lipidogram) osnovna je dijagnostička metoda za otkrivanje hiperlipidemije. Budući da je hiperlipidemija asimptomatska bolest, preporuča se provođenje probira na hiperlipidemiju, što se obično vrši tijekom sistematskih pregleda. Probirom moraju biti obuhvaćene sve osobe s povećanim rizikom od razvoja i postojanja hiperlipidemije te odrasle osobe bez obzira na prisutnost rizičnih čimbenika (≥ 40 godina za muškarce i ≥ 50 godina za žene). Uzorkovanje krvi za lipidogram preporuča se provesti nakon perioda gladovanja u trajanju od 12 do 14 sati. S obzirom na postojanje intraindividualnih varijacija u vrijednostima lipida (do 10 % za vrijednosti kolesterola i do 20 % za vrijednosti triglicerida), preporuča se provođenje dva uzorkovanja u razmaku od dva tjedna.
Klasični lipidogram uključuje mjerenje ukupnih triglicerida, kolesterola te LDL i HDL kolesterola. Trigliceridi, kolesterol te HDL kolesterol mjere se direktno dok se LDL izračunava računski (Friedewaldova formula: LDL = ukupni kolesterol – trigliceridi/5 - HDL), iako se u zadnje vrijeme i ta vrijednost dobiva direktnim mjerenjem. Referentne vrijednosti ovih parametara prikazane su u Tablici 8.15.
|
Tablica 8.15. Klasični lipidogram: preporučene vrijednosti
|
|
Ukupni trigliceridi
|
< 1,7 mmol/L
|
|
Ukupni kolesterol
|
< 5 mmol/L
|
|
LDL kolesterol
|
< 3 mmol/L
|
|
HDL kolesterol
|
> 1,0 mmol/L (M); > 1,5 mmol/L (Ž)
|
Prošireni lipidogram uključuje dodatna mjerenja kao što su određivanje ne-HDL kolesterola te apolipoproteina A i B. Ne-HDL kolesterol služi kao procjena ukupne količine atoregnih lipoproteina u plazmi (LDL, VLDL, IDL), a računa se tako da se od ukupnoga kolesterola oduzme vrijednost HDL kolesterola. Budući da je Apo B glavni apoprotein aterogenih lipiproteinskih čestica, njegova vrijednost dobro kolerira s njihovom koncentracijom u plazmi te su studije pokazale da je određivanje Apo B jednako određivanju LDL-a i ne-HDL-a u predviđanju kardiovaskularnih rizika. Trenutne smjernice preporučuju određivanje apoliproteina B kod osoba s povišenom vrijednosti triglicerida, sa šećernom bolesti, metaboličkim sindromom, pretilih, kod osoba s niskim vrijednostima LDL-a, tada može biti alternativa u terapijskom praćenju (ciljne vrijednosti < 65 nmol/L). Određivanje Apo A služi u procjeni koncentracije HDL kolesterola u plazmi, s obzirom na to da je Apo A glavni apoprotein HDL-a, iako treba imati na umu da jedna HDL čestica može sadržavati jednu do pet Apo A molekula.
Genetska analiza. Točno razlučivanje o kojoj se vrsti nasljedne hiperkolesterolemije radi na temelju molekularnih tehnika (određivanje mutacije) ima manju ulogu u kliničkoj praksi, s obzirom na isti terapijski ishod. Komercijalno dostupne analize uključuju analizu mutacija ApoE, ApoB, LDLR i PCSK9 gena.
Liječenje. Terapijski pristup bolesnicima s hiperlipidemijom temelji se na smanjenju LDL kolesterola kao glavnom terapijskom cilju, što se može postići različitim nefarmakološkim i farmakološkim mjerama. U Tablici 8.16. nalaze se ciljne vrijednosti LDL kolesterola koje se preporučuju u liječenju hiperlipidemije, a temelje se na ukupnom kardiovaskularnom riziku za što koristimo SCORE tablice.
|
Tablica 8.16. Ciljne vrijednosti LDL-a u liječenju hiperlipidemije
|
|
Kardiovaskularni rizik
|
Objašnjenje
|
Ciljna vrijednost
|
|
Vrlo visoki
|
Dijagnoza kardiovaskularne bolesti, šećerna bolest tip 2, šećerna bolest tip 1 s oštećenjem ciljnih organa, umjerena do teška kronična bolest bubrega, dijagnostičkim metodama (koronarografijom ili CD karotida) dokazana ateroskleroza (plak) na epikardijalnim arterijama ili karotidama, SCORE rezultat ≥ 10 %
|
< 1,4 mmol/L i/ili ≥ 50 % smanjenje od bazalnih vrijednosti
|
|
Visoki
|
Šećerna bolest tip 2 bez drugih KV čimbenika ili oštećenja ciljnih organa, značajno visoki pojedinačni čimbenici rizika trigliceridi > 8 mmol/L, LDL > 4,9 mmol/L, RR ≥ 180/110 mmHg, osobe s obiteljskom hiperkolesterolomijom, s umjerenim bubrežnim zatajenjem 30 - 59 ml/min, SCORE rezultat ≥ 5, a manji od 10 %
|
< 1,8 mmol/L i/ili ≥ 50 % smanjenje od bazalnih vrijednosti
|
|
Umjereni
|
SCORE rezultat ≥ 1, a < 5 %
|
< 2,6 mmol/L
|
|
Niski
|
SCORE rezultat < 1 %
|
< 3,0 mmol/L
|
SCORE tablice. Za procjenu kardiovaskularnoga rizika u praksi se najčešće koriste SCORE tablicama (engl. systemic coronary risk estimation) kojima se procjenjuje desetogodišnji rizik od smrti uzrokovane kardiovaskularnim bolestima. Korištenje SCORE tablica namijenjeno je naizgled zdravim bolesnicima koji do sada nisu oboljeli od kardiovaskularne bolesti i nemaju vrlo visok ili visok rizik od obolijevanja od kardiovaskularnih bolesti. Procjena rizika SCORE tablicama temelji se na dobi i spolu bolesnika, kao i prisutnosti i izraženosti najvažnijih rizičnih čimbenika, a to su pušenje, hiperkolesterolemija te vrijednost sistoličkoga krvnog tlaka. Na temelju poznavanja ovih parametara iz SCORE tablice se jednostavnim pregledom može odrediti desetogodišnji rizik izražen u postocima (Slika 8.3.). Ovisno o postotku procijenjenoga rizika, bolesnike se svrstava u jednu od skupina: 1) osobe s niskim ili umjerenim rizikom (SCORE < 5 %); 2) osobe s visokim rizikom (SCORE 5 – 9 %) te 3) osobe s vrlo visokim rizikom (SCORE > 10 %). Razlikuju se dvije vrste SCORE tablica: one koje se koriste u zemljama s niskim rizikom od obolijevanja od kardiovaskularnih bolesti te one koje se koriste u zemljama s visokim i vrlo visokim rizikom u koje ubrajamo i Hrvatsku.
Slika 8.3. SCORE tablica
Nefarmakološke mjere liječenja hiperlipidemije u prvom redu uključuju promjene prehrambenih navika, redovno provođenje tjelesne aktivnosti te uklanjanje i/ili kontrolu drugih rizičnih čimbenika (prestanak pušenja, smanjenje unosa alkohola, smanjenje prekomjerne tjelesne težine). Razina LDL kolesterola ne može se smanjiti za više od 10 % nefarmakološkim mjerama liječenja. U Tablici 8.17. su prikazani trenutno dostupni dokazi o utjecajima promjene načina života i prehrane na lipoproteine, naznačujući razinu učinka i dokaza u odnosu na utjecaje na određenu klasu lipoproteina.
|
Tablica 8.17. Utjecaj nefarmakoloških mjera liječenja hiperlipidemije
|
|
Intervencija
|
Razina učinka*
|
Razina dokaza
|
|
Promjene životnoga stila u snižavanju vrijednosti triglicerida i LDL kolesterola
|
|
Izbjegavanje unosa trans masti
|
++
|
A
|
|
Smanjenje unosa zasićenih masti
|
++
|
A
|
|
Povećanje unosa vlakana
|
++
|
A
|
|
Korištenje funkcionalne hrane obogaćene fitosterolima
|
++
|
A
|
|
Korištenje nutrijenata kvasca crvene riže
|
++
|
A
|
|
Smanjenje povećane tjelesne mase
|
++
|
A
|
|
Smanjenje unosa kolesterola prehranom
|
+
|
B
|
|
Povećanje tjelesne aktivnosti
|
+
|
B
|
|
Promjene životnoga stila u snižavanju vrijednosti trigliceridima bogatih lipoproteina
|
|
Smanjenje povećane tjelesne mase
|
+
|
A
|
|
Smanjenje unosa alkohola
|
+++
|
A
|
|
Povećanje tjelesne aktivnosti
|
++
|
A
|
|
Smanjenje unosa ukupne količine ugljikohidrata
|
++
|
A
|
|
Korištenje dodataka prehrani ω-3 polinezasićenih masti
|
++
|
A
|
|
Smanjenje unosa mono- i disaharida
|
++
|
B
|
|
Zamjena zasićenih masti s mono- ili polinezasićenim mastima
|
+
|
B
|
|
Promjene životnoga stila u povećanju vrijednosti HDL kolesterola
|
|
Izbjegavanje unosa trans masti
|
++
|
A
|
|
Povećanje tjelesne aktivnosti
|
+++
|
A
|
|
Smanjenje povećane tjelesne mase
|
++
|
A
|
|
Smanjene unosa ugljikohidrata i njihova zamjena nezasićenim mastima
|
++
|
A
|
|
Nastavak umjerenoga unosa alkohola (oni koji ga uzimaju)
|
++
|
B
|
|
Prestanak pušenja
|
+
|
B
|
* Interpretacija razine učinka: +++ (> 10 %); ++ ( 5 - 10 %), + (< 5 %)
Farmakološke mjere liječenja podrazumijevaju primjenu hipolipemika, lijekova koji svojim farmakološkim učinkom dovode do snižavanja vrijednosti lipida u krvi. U ovu skupinu lijekova ubrajamo statine, nikotinsku kiselinu, fibrate, ionske izmjenjivače, inhibitore apsorpcije kolesterola, PCKS9 inhibitore te riblje ulje. Primjena statina indicirana je kod osoba mlađih od 75 godina u svrhu primarne prevencije. U nastavku će biti opisane osnovne karakteristike pojedinih lijekova te njihova primjena ovisno o vrsti hiperlipidemije.
Statini su lijekovi koji inhibiraju HMG-CoA reduktazu, limitirajući enzim u procesu endogene biosinteze kolesterola. Posljedično inhibiciji toga enzima dolazi do pada količine kolesterola u stanicama zbog čega dolazi do kompenzatornoga odgovora u vidu povećanja ekspresije LDL receptora na površini stanica čime se povećava uklanjanje LDL-a i drugih lipoproteina koji sadržavaju Apo B na svojoj površini. Na taj se način dominantno snižava količina kolesterola u krvi, a u manjoj mjeri i triglicerida, dok se količina HDL kolesterola povećava što ove lijekove svrstava među najučinkovitije hipolipemike. Osim toga, statini imaju i druge pogodne učinke u prevenciji i liječenju kardiovaskularnih bolesti: antiaterogeni učinak, protuupalni učinak te antiagregacijski učinak. Brojne studije pokazale su pozitivan učinak statina u primarnoj i sekundarnoj prevenciji kardiovaskularnoga mortaliteta i morbiditeta, kao i pozitivan učinak na usporavanje i regresiju ateroskleroze koronarnih arterija (stabilizacija plakova). Najčešće nuspojave koje se opisuju kod ovih bolesnika uključuju umjereno povišenje jetrenih transaminaza, mialgije i porast kreatin kinaze (uz odsutnost drugih potencijalnih uzroka), konstipaciju, insomniju te rijetko pojavu rabdomiolize. Danas su na raspolaganju različiti statini: simvastatin (20 - 80 mg/dan), atorvastatin (10 - 80 mg/dan), pravastatin (20 - 80 mg/dan), fluvastatin (20 - 80 mg/dan), lovastatin (20 - 80 mg/dan) te rosuvastatin (10 - 40 mg/dan). Preporuča se dozu lijeka uzeti pred spavanje budući da je aktivnost HMG-CoA reduktaze najizraženija tijekom noći.
Derivati nikotinske kiseline dio su kompleksa vitamina B čiji farmakološki mehanizam kojime ostvaruju svoj hipolipemični učinak nije poznat, iako se smatra da se radi o smanjenoj endogenoj sintezi lipida u jetri. Ovi lijekovi dokazano smanjuju količinu triglicerida za 15 - 20 %, LDL kolesterola za 5 - 10 % te dovode do povišenja HDL kolesterola za 10 - 20 %. Početna doza kreće se od 100 do 500 mg, a maksimalna dnevna doza iznosi 1,5 do 2 grama. Nuspojave koje se mogu javiti kod bolesnika su iznenadni napadi crvenila i glavobolje, bolova u trbuhu, mučnine i povraćanja. Navedene neželjene nuspojave smanjuju se s vremenom, a na njihovo ublažavanje povoljno utječe primjena acetilsalicilne kiseline.
Fibrati su hipolipemici koji se primarno koriste u liječenju hipertrigliceridemije. Svoje učinke ostvaruju aktivacijom nuklearnoga receptora PPARα koji je uključen u regulaciju metabolizma lipida. Aktivacijom ovoga receptora ostvaruju se različiti učinci među kojima je najznačajniji pojačana aktivnost lipoprotein lipaze (povećana hidroliza triglicerida). Osim značajnijega sniženja razine triglicerida (25 - 35 %), u manjoj mjeri ostvaruje se i snižavanje vrijednosti LDL kolesterola (10 - 15 %) te povećanje vrijednosti HDL kolesterola (noviji lijekovi i do 50 %). Nije pokazan jasan učinak na smanjenje kardiovaskularnih događaja i smrtnosti. Fibrati se obično dobro podnose, a najčešće su nuspojave povećan rizik od razvoja žučnih kamenaca, prolazna miopatija (osobito ako se kombiniraju sa statinima) te povišenje jetrenih transaminaza. Najčešće korišteni fibrati su gemfibrozil (2 x 600 mg/dan) te fenofibrat (1 x 160 mg/dan).
Ionski izmjenjivači su hipolipemici koji svoj učinak ostvaruju vežući na sebe žučne kiseline u tankom crijevu sprječavajući njihovu reapsorpciju i enterohepatično kruženje. Na taj se način pojačano gube žučne kiseline što jetra nastoji kompenzirati pojačanom produkcijom novih, a kao osnovni izvor za sintezu žučnih kiselina koristi se kolesterol. Na taj se način potiče pojačana potrošnja kolesterola i njegovo lučenje iz organizma u obliku žučnih kiselina. Glavne nuspojave ionskih izmjenjivača su gastrointestinalne (bolovi u trbuhu, proljev, napuhanost, flatulencija). Zbog mogućnosti da na sebe veže i druge lijekove, preporuča se uzimanje drugih lijekova jedan sat prije i četiri sata nakon uzimanja ionskih izmjenjivača. U kliničkoj praksi primjenjuju se kolestiramin (početna doza 4 g, maksimalna 32 g/dan), kolestipol (početna doza 5 g, maksimalna 40 g/dan) te kolesevelam (početna doza 3750 mg, maksimalna 4375 mg/dan).
Inhibitori apsorpcije kolesterola svoj učinak ostvaruju kočenjem apsorpcije kolesterola iz crijeva. Najčešće se primjenjuje ezetimib koji inhibira NPC1L1 protein koji služi kao transportna molekula koja omogućava prijenos kolesterola u enterocite. Osim toga, smanjivanjem egzogenoga unosa kolesterola pojačava se ekspresija LDL receptora na površini stanica kako bi se nadoknadio manjak kolesterola u stanicama te se i na taj način pojačano uklanja LDL iz krvi. Kao i kod ionskih izmjenjivača, najčešće nuspojave vezane su uz probavni sustav. Ezetimib se koristi jednom na dan u dozi od 10 mg. Obično se dodaje statinima u liječenju hiperkolesterolemije kao druga linija terapije s obzirom da je dokazan i povoljan učinak na kardiovaskularne događaje i smrtnost.
PCKS9 inhibitori (evolocumab, alirokumab) predstavljaju noviju skupinu hipolipemika čiji se učinak ostvaruje inhibicijom PCKS9 enzima koji sudjeluje u degradaciji LDL receptora s površine stanice. Njegovom inhibicijom sprečava se degradacija LDL receptora te se oni duže zadržavaju na površini stanica što rezultira povećanim uklanjanjem LDL čestica iz krvi. Radi se o monoklonalnim antitijelima koja se primjenjuju supkutano, obično dva puta ili jednom mjesečno, ovisno o dozi. Kliničke studije koje su provedene pokazuju dobru učinkovitost u postizanju snižavanja vrijednosti LDL kolesterola (50 - 70 %), bez većega utjecaja na HDL kolesterol i trigliceride, kao i smanjenje kardiovaskularnih događaja, no bez značajnoga učinka na kardiovaskularnu smrtnost, vjerojatno zbog kratkoga vremenskog trajanja studija. Najčešće prijavljene nuspojave su svrbež na mjestu aplikacije te simptomi nalik gripi. Neke studije su pokazale i utjecaj na kognitivne funkcije što poziva na oprez prilikom davanja lijeka.
Omega-3 masne kiseline (eikosapentaenoična kiselina – EPA, dokosaheksaenoična kiselina – DHA) dokazano utječu na smanjenje vrijednosti tiriglicerida, ali je njihov učinak minimalan. Mehanizam djelovanja je slabo poznat, a smatra se da utječu na PPARα te smanjeno lučenje Apo B. Podaci o učinku na kardiovaskularne događaje i smrtnost ostaju neuvjerljivi, a čini se da je klinička učinkovitost omega-3 masnih kiselina povezana s njihovim nelipidnim učincima. Preporučena dnevna doza iznosi 4 g. Najčešće nuspojave su dispneja i zadah po ribi (navedene kiseline dobivaju se iz ribljega ulja).
Terapijske smjernice za liječenje hiperlipidemije. U nastavku teksta biti će predstavljene preporuke Europskoga kardiološkog društva (izv. European Society of Cardiology, ESC) te Europskog društva za aterosklerozu (izv. European Atherosclerosis Society, EAS) za liječenje hipertrigliceridemije i hiperkolesterolemije iz 2019. godine. Mnoga klinička ispitivanja jasno su pokazala da što su niže postignute vrijednosti LDL kolesterola, to je niži rizik od budućih kardiovaskularnih događaja, no bez donje granice za vrijednosti LDL kolesterola ili učinka „J’-krivulje“. Uz to, studije kliničke sigurnosti ovih vrlo nisko postignutih vrijednosti LDL-a pokazuju dobar sigurnosni profil, iako je potrebno praćenje tijekom duljega razdoblja. Za povišenje HDL kolesterola nedavne su studije pokazale da trenutno dostupne terapije ne smanjuju rizik od aterosklerotske kardiovaskularne bolesti (ASKVB). Konačno, humane studije Mendelove randomizacije pokazale su kritičnu ulogu LDL kolesterola i drugih lipoproteina koji sadrže ApoB, sadržane u kolesterolu, u nastanku aterosklerotskih plakova i srodnim kasnijim kardiovaskularnim događajima. Dakle, više ne postoji ‘hipoteza o LDL kolesterolu’, već su utvrđene činjenice da su povećane vrijednosti LDL kolesterola uzročno povezane s ASKVB-om, te da snižavanje LDL-čestica i drugih lipoproteina koji sadrže ApoB smanjuje kardiovaskularne događaje.
Preporuke za liječenje hiperkolesterolemije uključuju sljedeće: 1) dati statin u najvišoj dozi ili najvećoj dozi koju bolesnik podnosi kako bi se postigle ciljne vrijednosti LDL kolesterola; 2) ako ciljne vrijednosti nisu postignute uz statin u najvišoj dozi, odnosno najvećoj podnošljivoj dozi, dodati ezetimib 10 mg; 3) kod bolesnika s vrlo visokim KV rizikom i bolesnika s heterozigotnom porodičnom hiperkolesterolemijom koji ne postižu ciljne vrijednosti LDL-a unatoč najvećim dopuštenim dozama statina u kombinaciji s inhibitorima apsorpcije kolesterola, ezetimibom, preporuča se uvođenje PCSK9 inhibitora.
Preporuke za liječenje hipertrigliceridemije uključuju sljedeće: 1) primjena farmakoterapije u liječenju hipertrigliceridemije preporuča se kod visoko rizičnih bolesnika s vrijednostima triglicerida većim od 2,3 mmol/L; 2) statini se preporučavaju kao lijek prvoga izbora za smanjenje KV rizika kod visoko rizičnih bolesnika s hipertrigliceridemijom (uz dodatak omega-3 masnih kiselina 2 x 2 gr); 3) kod visoko rizičnih bolesnika s trigliceridima višim od 2,3 mmol/L unatoč primjeni statina može se dodatno primijeniti fenofibrat u kombinaciji sa statinima.
Praćenje bolesnika. Nakon uvođenja terapije kontrolni lipidogram preporuča se nakon 8 (±4) tjedana, a ako su postignute ciljne vrijednosti, dalje se preporučuje 1 - 2 godišnje. Ako nisu postignute ciljne vrijednosti i korigirana je terapija, sljedeće mjerenje preporuča se nakon 8 (+4) tjedana, kao i nakon svake sljedeće promjene doze i/ili lijekova. Ako se radi o bolesnicima koji preboljeli akutni koronarni sindrom, obavezna je kontrola nakon 4 - 6 tjedana. Po istom principu (nakon osam tjedana) preporuča se i kontrola jetrenih transaminaza. Porast jetrenih transaminaza do tri puta u odnosu na normalne vrijednosti ne zahtijeva korekciju terapije, no ako se oni povise za više od tri puta od normalnih vrijednosti, preporuča se smanjenje doze ili prekid uzimanja statina. Kontrola jetrenih transaminaza, ako su povišene, provodi se za 4 do 6 tjedana. Redovne kontrole kreatin kinaze (CK) nisu potrebne osim u slučaju kliničkih manifestacija (mialgije).
POREMEĆAJI METABOLIZMA MOKRAĆNE KISELINE
Uvod. Mokraćna ili urična kiselina predstavlja konačni produkt katabolizma (razgradnje) purina u ljudskom organizmu. Purini (adenin, gvanin) su dušične baze koje imaju važnu ulogu u osnovnim fiziološkim procesima među kojima je najbitnija izgradnja nukleinskih kiselina (DNA i RNA). Oni zajedno sa šećerom (riboza) i fosfatnom skupinom formiraju nukleotid, osnovni gradivni element nukleinskih kiselina. Purini u organizmu u najvećem dijelu nastaju de novo sintezom, ali se u znatnoj mjeri unose i putem hrane te mogu nastati resintezom iz razgrađenih nukleotida. Put razgradnje purinskih baza do mokraćne kiseline posredovan je različitim enzimima i međuproduktima, od kojih su najbitniji hipoksantin i ksantin te enzim ksantin-oksigenaza (Slika 8.4.). Mokraćna kiselina je slaba kiselina koja se u tjelesnim tekućinama nalazi u ioniziranom obliku (mononatrijev urat) koji u stanjima prezasićenosti može stvarati kristalne strukture (kristalizacija). Najveći dio urata izlučuje se mokraćom putem bubrega, a manji se dio izlučuje i stolicom preko crijeva. Osnovni poremećaji metabolizma mokraćne kiseline su hiperuricemija i hipouricemija.
Slika 8.4. Metabolizam mokraćne kiseline u organizmu
(KO – ksantin-oksigenaza)
Glavni ekskrecijski put za izlučivanje mokraćne kiseline su bubrezi. Ona se najprije filtrira u glomerulima (glomerularna filtracija), zatim se znatan dio reapsorbira u tubulima (tubularna reapsorpcija) te ponovno aktivno secernira u mokraću (tubularna sekrecija), a sve to je uvjetovano različitim specifičnim transporterima (URAT1, OAT1, OAT2, OAT3, OAT4).
HIPERURICEMIJA
Definicija. Hiperuricemija se definira kao stanje povećane koncentracije mokraćne kiseline (urata) u krvi i to u vrijednostima višima od 415 µmol/L za muškarce i 360 µmol/L za žene. Radi o relativno čestom patološkom laboratorijskom nalazu te se smatra da 5 - 10 % osoba u općoj populaciji ima povećane vrijednosti urata u krvi, bilo da se radi o simptomatskoj, bilo o asimptomatskoj hiperuricemiji.
Etiologija. Hiperuricemija može nastati zbog pojačanoga stvaranja ili smanjenoga izlučivanja urata, njihove kombinacije, a bitnu ulogu ima i pojačan unos purina putem hrane. Ovisno o tome radi li se o nasljednom ili stečenom poremećaju razlikujemo primarnu (nasljednu) i sekundarnu (stečenu) hiperuricemiju.
Pojačana produkcija urata povezana je s pojačanom endogenom sintezom purinskih baza, što može biti genetski uvjetovano ili se može javiti sekundarno uslijed pojačane stanične proliferacije ili razgradnje (Tablica 8.18.). Pojačano stvaranje purinskih baza nalaže i njihovu pojačanu razgradnju što rezultira hiperuricemijom.
|
Tablica 8.18. Uzroci pojačane produkcije purina
|
|
Primarni uzroci hiperprodukcije purina
|
Sekundarni uzroci hiperprodukcije purina
|
|
Deficijencija hipoksantin-guanin-fosforiboziltransferze
Hiperaktivnost sintetaze fosforibozilpirofosfata
|
Mijeloproliferativne i limfoproliferativne bolesti
Hemoliza, rabdomioliza, citostatska terapija
|
Smanjeno izlučivanje urata najčešći je patogenetski mehanizam nastanka hiperuricemije, a može biti posljedica smanjene glomerularne filtracije (kronična bolest bubrega) ili poremećaja transporta urata u tubulima (pojačana reapsorpcija, smanjena sekrecija) što može biti genetski uvjetovano (poremećaji transportera) ili se može javiti sekundarno uslijed bolesti koje zahvaćaju tubule ili prisutnosti molekula koje interferiraju s transportom urata (Tablica 8.19.).
|
Tablica 8.19. Najčešći uzroci smanjenoga izlučivanja urata
|
|
Kronična bubrežna bolest
Policistična bolest bubrega
Dijabetes insipidus
Metabolička acidoza
|
Toksemija u trudnoći
Hipotireoza
Sarkoidoza
Hipertenzija
|
Salicilati
Diuretici
Alkohol
Levedopa
|
Utjecaj prehrane. Unos egzogenih purina putem hrane može značajno utjecati na vrijednosti urata u krvi te pridržavanje restrikcija unosa hrane bogate purinima dovodi do značajnoga sniženja vrijednosti serumskih urata, kao i do smanjenja sekrecije putem urina. Namirnice bogate purinima su meso i životinjske masnoće, iznutrice (jetra, bubrezi, mozak), slane morske ribe, maslac, slatko vrhnje, čokolada te određeno povrće (grah, grašak, špinat, šparoge, mahune, kelj) i voće (breskve, grožđe, marelice).
Patogeneza. Glavne patogenetske posljedice hiperuricemije povezane su s mogućnošću kristalizacije soli urata i nastanka kristala koji se mogu taložiti u različita tkiva i organe i uzrokovati njihova oštećenja, a najčešća takva stanja su urički artritis (giht), urička nefrolitijaza te urička nefropatija.
Klinička slika. Hiperuricemija se može prezentirati kao asimptomatski patološki laboratorijski nalaz ili se može očitovati nekom od svojih karakterističnih manifestacija (giht, nefrolitijaza). Danas se sve više govori i o povezanosti hiperuricemije s metaboličkim sindromom (63 % bolesnika s metaboličkim sindromom ima hiperuricemiju) što je posljedica djelovanja hiperinzulinemije na smanjenju ekskreciju urata putem bubrega. Negativni učinak urata na endotelnu funkciju pridonosi aterogenezi i smatra se bitnim rizičnim čimbenikom u razvoju kardiovaskularnih bolesti. Pojedini klinički entiteti opisani su u odgovarajućim dijelovima udžbenika (nefrologija, reumatologija).
Dijagnoza. Osnovni dijagnostički korak je laboratorijsko određivanje vrijednosti urata u serumu (do 415 µmol/L muškarci, do 360 µmol/L žene) te u 24-satnom urinu (3,6 - 5,4 mmol). Interpretaciju dobivenih nalaza treba staviti u kontekst kliničke slike te provesti druge, odgovarajuće dijagnostičke metode.
Liječenje. Osnovu liječenja čini prehrana bez purina te primjena lijekova koji smanjuju koncentraciju urata u krvi, a algoritam liječenja ovisi o kliničkim manifestacijama (asimptomatska hiperuricemija, giht, nefrolitijaza).
Antiurici su lijekovi koji se koriste u liječenju hiperuricemije, a u tu se svrhu primjenjuju lijekovi iz grupe inhibitora ksantin-oksigenaze (alopurinol, febuksostat). Smanjivanjem djelovanja enzima ksantin-oksigenaze smanjuje se pretvorba hipoksantina i ksantina u mokraćnu kiselinu čime se smanjuju vrijednosti urata u serumu. Detaljnije je o indikacijama i dozama ovih lijekova u specifičnim kliničkim stanjima pisano u odgovarajućim dijelovima udžbenika.
Asimptomatska hiperuricemija ne zahtijeva medikamentnu terapiju, nego se bolesnicima preporuča ustezanje od hrane bogate purinima. Jedina indikacija za primjenu antiurika u asimptomatskoj hiperuricemiji je stanje nakon agresivne kemoterapije, u svrhu prevencije nastanka sindroma lize tumora. Bolesnike kod kojih se slučajno otkrije asimptomatska hiperuricemija treba češće kontrolirati u smislu uočavanja i liječenja ostalih sastavnica metaboličkoga sindroma (hiperlipidemija, hipertenzija, hiperglikemija).
HIPOURICEMIJA
Hipouricemija se definira kao stanje smanjene koncentracije mokraćne kiseline (urata) u serumu i to u vrijednostima ispod 120 µmol/L. Ona može nastati kao posljedica smanjenoga stvaranja urata, pojačane ekskrecije urata ili njihovom kombinacijom. Hipouricemija je u većini slučajeva posljedica pojačanoga lučenja urata putem urina, a tome pridonose lijekovi (aspirin, losartam, fenofibrat), radiološka kontrastna sredstva, hiperalimentacija u sklopu totalne parenteralne prehrane, kao i neke bolesti i stanja (ciroza jetre, defekti tubularnoga transporta i sl.). To je rijetko stanje koje se susreće kod manje od 0,2 % osoba u općoj populaciji i 0,8 % u populaciji hospitaliziranih osoba. Hipouricemija nema veći klinički značaj i ne zahtijeva nikakve terapijske intervencije.
POREMEĆAJI METABOLIZMA PORFIRINA (PORFIRIJE)
Uvod. Porfirini predstavljaju skupinu molekula koje nastaju kao intermedijarni spojevi (međuprodukti) u procesu sinteze hema, sastavnoga dijela hemoglobina. Oni su građeni od četiri pirolna prstena, a sinteza svakoga pirolnog prstena kreće od prekursora glicina i molekule sukcinil-CoA. Cijeli put biosinteze hema sastoji se od više koraka u što je uključeno mnoštvo enzima pri čemu nastaju različiti međuprodukti (porfirini). Porfirije su skupina rijetkih i nasljednih bolesti koje karakteriziraju poremećaji specifičnih enzima u procesu biosinteze hema s posljedičnim nakupljanjem određenih porfirina u tijelu koji dovode do patoloških reakcija. U shematskom prikazu na Slici 8.5. prikazan je put biosinteze hema sa specifičnim enzimima i poremećajima koji nastaju uslijed poremećaja tih enzima.
|
Glicin + sukcinil-CoA
|
|
|
|
|
ALA-sintetaza
|
X-vezana sideroblastična anemija
|
|
Aminolevulinska kiselina (ALA)
|
|
|
| |
ALA-dehidrataza
|
Plumboporfirija
|
|
Porfobilinogen (PBG)
|
|
|
| |
Hidroksimetilbilan-sintetaza
|
Akutna intermitentna porfirija
|
|
Hidroksimetilbilan
|
|
|
| |
Uroporfirinogen sintetaza
|
Kongenitalna eritropoetska porfirija
|
|
Uroporfirinogen III
|
|
|
| |
Uroporfirinogen dekarboksilaza
|
Porfirija kutanea tarda
|
|
Koproporfirinogen III
|
|
|
|
|
Koproporfirinogen oksidaza
|
Hereditarna koproporfirija
|
|
Protoporfirinogen IX
|
|
|
| |
Protoporfirinogen oksidaza
|
Porfirija variegata
|
|
Protoporfirin IX
|
|
|
| |
Ferokelataza
|
Eritropoetska protoporfirija
|
|
Hem
|
|
|
Slika 8.5. Shematski prikaz biosinteze hema i prikaz poremećaja metabolizma porfirina (profirije)
Ovisno o mjestu primarne hiperprodukcije i nakupljanja porfirina, porfirije se dijeli u tri osnovne skupine: eritropoetske porfirije (stanice crvene loze), hepatične porfirije (jetra) te hepatoeritropetske porfirije. Tri najčešća klinička oblika ovih poremećaja su: akutna intermitentna porfirija, porfirija kutanea tarda te eritropetska protoporfitija.
AKUTNA INTERMITENTNA PORFIRIJA
Definicija. Akutna intermitentna porfirija je rijedak nasljedni poremećaj metabolizma porfirina, a nastaje uslijed deficijencije enzima hidroksimetilbilan-sintetaze. Radi se o najčešćem obliku porfirija s prosječnom učestalošću od oko 5:100 000 u općoj populaciji. Bolest se nešto češće javlja kod žena u odnosu na muškarce.
Etiopatogeneza. Akutna intermitentna porfirija nastaje kao posljedica mutacije HMBS gena koji se nalazi na 11. kromosomu (11q23.3), a prenosi se autosomno-dominantnim putem. Posljedično ovoj mutaciji nalazimo manjkavost enzima hidroksimetilbilan-sintetaze zbog čega dolazi do patološkoga nakupljanja porfobilinogena, a do danas je opisano više od 300 mutacija ovoga gena. Homozigoti imaju apsolutni manjak ovoga enzima i bolest ispoljavaju rano u djetinjstvu, dok heterozigoti imaju smanjenu aktivnost enzima koja postaje simptomatska u odrasloj dobi. Kod većine bolesnika smanjena aktivnost enzima dovoljna je za odvijanje ovoga metaboličkog puta u fiziološkim uvjetima, no u stanjima njegove pojačane aktivnosti dolazi do izražaja manjkavosti enzima i pojave bolesti. To se obično događa u uvjetima pojačane stimulacije enzima ALA-sintetaze koji je ujedno i limitrajući enzim puta biosinteze hema, a ta enzimatska stimulacija može biti potaknuta nekim lijekovima, hormonima ili prehranom (smanjen unos kalorija i ugljikohidrata). Činjenica da bolest rijetko postaje simptomatska prije puberteta ukazuje na velik utjecaj spolnih hormona u aktivnosti ovoga metaboličkog puta. Pojačano nakupljanje porfobilinogena u tkivima dovodi do njegova toksičnog učinka i razvoja karakteristične kliničke slike. U akutnoj intermitentnoj porfiriji najčešće su zahvaćeni jetra, organi probavne cijevi (posebice crijeva) te živčani sustav.
Klinička slika. Manifestacija bolesti obično započinje u ranoj odrasloj dobi (treće desetljeće), a očituje se intermitentnom pojavom simptoma nakon izlaganja provocirajućim čimbenicima (lijekovi, hormonski pripravci, smanjen kalorijski unos) koji traju nekoliko dana te se potom povlače u potpunosti. Napadaji su obilježeni visceralnom i neurološkom simptomatologijom.
Visceralna simptomatologija. Bol u trbuhu je najčešći simptom akutne intermitentne porfirije, a obično je tup i slabo lokaliziran, rijetko grčevit. Bol mogu pratiti mučnina, povraćanje te pojava proljeva ili opstipacije. Mogući je i razvoj paralitičkoga ileusa s izraženom distenzijom crijevnih vijuga te se često pogrešno tumači kao kirurški akutni abdomen.
Neurološka simptomatologija može biti posljedica disfunkcije autonomnoga živčanog sustava (simpatička hiperreaktivnost), perifernoga živčanog sustava (periferna neuropatija) te središnjega živčanog sustava. Znakovi simpatičke hiperreaktivnosti su uz bol u trbuhu najstalniji znaci ovoga poremećaja, a uključuju pojavu tahikardije, hipertenzije, finoga tremora te pojačanoga znojenja. Periferna neuropatija obično zahvaća motorička vlakna te se očituje slabošću proksimalnih mišića udova (češće ruku). Rijetko, mišićna slabost može zahvatiti respiratorne mišiće i dovesti do respiratorne insuficijencije. Simptomi središnjega živčanog sustava poglavito se očituju psihičkim smetnjama (anksioznost, depresija, paranoja, nesanica).
Dijagnoza. Osnovni korak u dijagnostici ove bolesti je postavljanje sumnje na nju, što se treba temeljiti na osnovnim anamnestičkim podacima: abdominalne tegobe nejasne etiologije koje spontano prolaze, povezanost s uzimanjem određenih lijekova ili gladovanjem, pojava simptoma simpatičke hiperreaktivnosti i dr. Sama dijagnoza se postavlja mjerenjem porfobilinogena u urinu, mjerenjem aktivnosti enzima hidroksimetilbilan-sintetaze te genetskom analizom mutacija HMBS gena.
Određivanje porfirina u urinu i stolici. Pri akutnoj intermitentnoj porfiriji u urinu nalazimo povišene vrijednosti porfobilinogena (50 - 200 mg/24 sata, normalno 0 - 4 mg/24 sata) te aminolevulinske kiseline (20 - 100 mg/24 sata, normalno 1 - 7 mg/24 sata). Vrijednosti su najizraženije u vrijeme akutnoga napadaja, a po smirivanju simptoma i njihove se vrijednosti postupno smanjuju, iako kod nekih pojedinaca mogu ostati povišene i između napadaja. Porfirini u stolici su uredni ili minimalno povišeni.
Određivanje enzima hidroksimetilbilan-sintetaze u perifernim eritrocitima ukazuje na njihovu sniženu razinu što ima visok dijagnostički značaj, ali treba imati na umu da razine ovoga enzima u eritrocitima mogu biti uredne unatoč aktivnoj bolesti (mutacije koje pogađaju samo neeritrocitne enzime; povišene vrijednosti u uvjetima stimulirane eritropoeze i sl.).
Genetska analiza i dokazivanje specifične mutacije HMBS gena danas je zlatni standard za postavljanje konačne dijagnoze ove bolesti i treba se provesti kod svih bolesnika sa suspektnom kliničkom slikom i/ili patološkim biokemijskim pretragama.
Liječenje. Bolesnike s akutnim napadajem bolesti potrebno je bolnički liječiti. Liječenje podrazumijeva simptomatske i specifične mjere. Simptomatsko liječenje uključuje kontrolu bola (narkotični analgetici), odgovarajuću hidraciju i ispravak elektrolitnoga disbalansa, a za kontrolu neuroloških simptoma preporuča se primjena kratkodjelujućih benzodiazepina u niskim dozama. Prilikom primjene lijekova svakako treba paziti da oni ne pogoršavaju akutnu intermitentnu porfiriju (primjeri nekih lijekova navedeni su u Tablici 8.20.). Specifično liječenje podrazumijeva primjenu glukoze i hema.
Liječenje glukozom osnovni je terapijski postupak budući da glukoza inhibira ALA-sintetazu te tako smanjuje simptome bolesti. Primjenjuje se 300 - 500 g/dan i to peroralnim putem ako bolesnik može jesti ili intravenski ako ne može. Ova terapija dostatna je za srednje izražene napadaje.
Liječenje hemom rezervirano je za teže kliničke oblike, a temelji se na inhibiciji ALA-sintetaze. Liječenje treba započeti nakon postavljanja dijagnoze (ili nakon provedenoga uzorkovanja), a preporučena doza hema iznosi 3 - 4 mg/kg kroz četiri dana. Primjenjuje se intravenski, a postoji više pripravaka (hidroksihem, hem arginat).
Sve bolesnike potrebno je podučiti o naravi bolesti te savjetovati izbjegavanje provocirajućih čimbenika, posebice oprez kod uzimanja određenih lijekova (provjera kako utječu na bolest) te izbjegavanje gladovanja.
|
Tablica 8.20. Lijekovi u akutnoj intermitentnoj porfiriji
|
|
Sigurni
|
Paracetamol, acetazolamid, alopurinol, amilorid, ACE inhibitori, antagonisti aldosteronskih receptora, aspirin, atropin, beta blokatori, bromidi, cimetidin, eritropetin, gentamicin, inzulin, glukokortikoidi, opioidni analgetici, ofloksacin, penicilinski antibiotici, ranitidin, tetraciklin
|
|
Nesigurni
|
Barbiturati, blokatori kalcijskih kanala, karbamazepin, klonazepam, danazol, diklofenak i drugi NSAID-i, ergotamini, mebrobamat, metoklopramid, fenitoin, pirazinamid, rifampicin, sulfonamidi, valporatna kiselina,
|
Prognoza. Ako se bolest prepozna na vrijeme i pravilno se zbrinu akutni napadaji, oni prolaze brzo i bez trajnih posljedica. Učestali napadaji mogu rezultirati trajnim neurološkim promjenama.
PORFIRIJA KUTANEA TARDA
Definicija. Porfirija kutanea tarda označava poremećaj metabolizma porfirina koji nastaje uslijed deficijencije enzima uroporfirinogen dekarboksilaze u jetri, a karakteristično se manifestira kožnim lezijama. Za razliku od ostalih porfirija, ovaj oblik se javlja češće kod muškaraca u odnosu na žene.
Etiopatogeneza. Bolest se razvija uslijed manjkavosti enzima uroporfirinogen dekarboksilaze u jetri (< 20 % normalne aktivnosti), a što se može razviti sporadično (tip I) ili može biti nasljedno uvjetovano (tip II). Obiteljski tip (tip II) nastaje kao posljedica mutacije UROD gena koji se nalazi na 1. kromosomu (1p34.1) i prenosi se autosomno-recesivnim putem, dok kod sporadičnoga oblika (tip I) ne nalazimo mutacije navedenoga gena. Kod tih se osoba smanjena aktivnost enzima povezuje s kroničnim alkoholizmom, pušenjem, estrogenskom terapijom, virusnim infekcijama (HVC, HIV) te mutacijama HFE gena i povišenim razinama željeza u serumu. Simptomi bolesti posljedica su pojačanoga stvaranja i nakupljanja uroporfirinogena III, tj. njegovih metabolita (uroporfirin, hepta-, penta- i heksakarboksiporfirin, isokoproporfirin) u raznim tkivima i organima, a osobito u koži gdje imaju sposobnost apsorpcije svjetlosti u ultraljubičastom području što ih električki podražuje te postaju kemijski reaktivni spojevi koji ulaze u spojeve s okolnim strukturama dovodeći do njihova oštećenja.
Klinička slika. Osnovna klinička manifestacija ovoga oblika porfirije je kožna fotoosjetljivost. Ona se ispoljava na dijelovima kože koji su izloženi sunčevim zrakama (ruke, noge, lice), a očituje se pojavom vezikula i bula koje kasnije pucaju te nastaju rane prekrivene krustama koje sporo cijele uz nastanak ožiljaka. Ponekad se javlja hiperpigmentacija i hipertrihoza na pogođenim mjestima, a moguće su i kalcifikacije što uz ožiljke daje sliku nalik sklerodermiji (pseudosklerodermija). Zahvaćenost jetre obično se očituje samo povišenim vrijednostima jetrenih transaminaza te ponekad hepatomegalijom, a rizik od nastanka ciroze i hepatocelularnoga karcinoma raste s duljinom trajanja bolesti. Neurološki simptomi u ovom obliku porfirije nisu evidentirani.
Dijagnoza se temelji na kliničkom nalazu te poremećaju specifičnih biokemijskih nalaza, a potvrđuje se genetskom analizom. Biopsija jetre indicirana je u nekim slučajevima bolesti. U općim laboratorijskim nalazima obično nalazimo povišene jetrene transaminaze te povećane vrijednosti serumskoga željeza.
Određivanje porfirina u urinu i stolici. Karakteristično u ovom obliku porfirija nalazimo povišene vrijednosti uroporfirina i heptakarboksiporfirina u urinu, a mogu se naći i povišene razine aminolevulinske kiseline uz uredne vrijednosti porfobilinogena (razlikovanje od akutne intermitentne porfirije gdje je i porfobilinogen povišen) te drugih metabolita uroporfirinogena III (heksakarboksiporfirin, pentakarboksiporfirin). U stolici se mogu naći povišene vrijednosti isokoproporfirina.
Određivanje enzima uroporfirinogen dekarboksilaze u perifernim eritrocitima, jetri ili kulturi kožnih fibroblasta služi za razlikovanje pojedinih podvrsta ove porfirije (tip II pokazuje značajno sniženje vrijednosti navedenoga enzima, < 50 % normalnih vrijednosti).
Genetska analiza i otkrivanje specifičnih mutacija UROD gena omogućava definitivno razgraničenje između bolesti tip I i tip II. Do danas je poznato više od 200 mutacija ovoga gena, a one se nađu kod svega 20 % bolesnika, dok ostalih 80 % čini sporadični oblik bolesti (tip I).
Liječenje. Osnovu liječenje čine preventivne mjere, odnosno izbjegavanje provocirajućih čimbenika (oprez kod primjene određenih lijekova, izbjegavanje hormonskih pripravaka, prestanak unosa alkohola i pušenja) te mjere smanjenja vrijednosti serumskoga željeza (povremene venepunkcije ili primjena vezača željeza kao što je deferoksamin). Ako se kožne promjene ne povlače ili recidiviraju unatoč navedenim mjerama, u terapiju se može dodati hidroksiklorokin (100 mg) ili klorokin (150 mg) dva puta tjedno kroz nekoliko mjeseci. Točan mehanizam učinaka ovih lijekova je nepoznat.
Prognoza bolesti uz pridržavanje izbjegavanja provocirajućih čimbenika kod većine bolesnika je dobra. Česti recidivi mogu dovesti do kroničnih kožnih promjena (pseudosklerodermija).
ERITROPOETSKA PROTOPORFIRIJA
Definicija. Eritropetska protoporfirija je treći najčešći oblik porfirija koji se obično javlja u djetinjstvu, a nastaje uslijed deficijencije enzima ferokelataze, zadnjega enzima u metaboličkom putu biosinteze hema, koji pretvara protoporfirin IX u hem.
Etiopatogeneza. U 90 % slučajeva radi se o autosomno-recesivnom nasljednom poremećaju u kojemu nalazimo mutaciju FECH gena (18q21.3) koji je odgovoran za nastanak ferokelataze, dok se u 10 % slučajeva ta mutacija ne nalazi. Posljedično manjkavosti navedenoga enzima dolazi do pojačane produkcije protoporfirina u eritrocitima i jetri koji potom ulaze u krv, talože se u koži te se izlučuju putem žuči i fecesa. Lučenjem putem žuči dovodi do taloženja i kristalizacije s posljedičnom sklonosti razvoju konkramenata i kolestaze.
Klinička slika. Bolest se ponajprije manifestira kožnim simptomima koji se razlikuju od ostalih porfirija, a pojavljuju se već u ranom djetinjstvu. Promjene se pojavljuju nekoliko minuta nakon izlaganja kože sunčevom svjetlu u vidu edema, eritema i svrbeža. Opisane promjene na koži glave mogu nalikovati angioedemu. Za razliku od porfirije kutanee tarde pojava vezikula i bula iznimna je rijetkost te se promjene povlače bez stvaranja trajnih ožiljaka. Od ostalih kliničkih manifestacija bilježi se umjerena mikrocitna anemija, povećana učestalost žučnih kamenaca koji sadrže protoporfirin te se u manje od 5 % slučajeva razvija protoporfirinska hepatopatija koju obilježava izražena kolestaza te brzo napredovanje prema fibrozi i cirozi.
Dijagnoza se postavlja na osnovi kliničkoga nalaza, specifičnih biokemijskih nalaza te genetskoga testiranja. Specifični biokemijski poremećaji uključuju povećanu razinu protoporfirina u eritrocitima i stolici uz uredne vrijednosti porfirina u urinu. Pri određivanju protoporfirina u eritrocitima razlikujemo dvije frakcije: slobodnu i vezanu za cink što je bitno u diferencijalnoj dijagnozi eritropoetske protoporfirije spram drugih stanja koja mogu dovesti do povećanih vrijednosti protoporfirina u eritocitima (trovanje olovom, sideropenija, hemoliza), s obzirom na to da je u tim stanjima povišena frakcija protoporfirina vezana za cink. Dijagnoza se dodatno potvrđuje mjerenjem aktivnosti ferokelataze u limfocitima ili kulturama kožnih fibroblasta (snižena) te molekularnim metodama dokazivanja specifičnih mutacija FECH gena.
Liječenje. Osnovu liječenja čini izbjegavanje izlaganja sunčevoj svjetlosti uz zaštitu kože odjećom ili kremama s visokim zaštitnim čimbenikom. Peroroalna primjena β-karotena može poboljšati toleranciju kože na sunčevu svjetlost. U liječenju protoporfirinske hepatopatije primjenjuje se kolestiramin (protoporfirinski adsorbens), vitamin E (snažan antioksidans) uz ostale ustaljene metode liječenja.
DIFERENCIJALNA DIJAGNOZA PORFIRIJA
Inicijalna dijagnostička pretraga kod sumnje na porfiriju je određivanje porfirina u urinu. Na temelju nalaza pojedinih (najznačajnijih) porfirina u urinu može se orijentacijski zaključiti o kojoj se vrsti porfirije radi. Diferencijalna dijagnoza porfirija na temelju porasta sadržaja pojedinih metabolita u 24-satnom uzorku urina prikazana je u Tablici 8.21.
|
Tablica 8.21. Diferencijalna dijagnoza porfirije
|
|
Tip porfirije
|
U
|
7
|
6
|
5
|
CI
|
CIII
|
|
Porfirija kutanea tarda
|
↑↑
|
↑↑
|
↑
|
↑
|
↑
|
↑
|
|
Akutna intermitentna porfirija
|
↑↑
|
↑
|
↑
|
↑↑
|
↑
|
↑↑
|
|
Porfirija variegata
|
↑↑
|
↑
|
↑
|
↑↑
|
↑
|
↑↑
|
|
Hereditarna koproporfirija
|
↑
|
↑
|
↑
|
↑↑
|
N
|
↑↑
|
|
Eritropoetska protoporfirija
|
V
|
N
|
N
|
V
|
↑
|
V
|
|
Kongenitalna eritropoetska porfirija
|
↑↑
|
↑
|
↑
|
↑
|
↑
|
--
|
|
Deficijencija porfobilinogen sintetaze
|
↑
|
↑
|
↑
|
↑↑
|
--
|
↑↑
|
Kratice: U (uroporfirin), 7 (heptakarboksiporfirin), 6 (heksakarboksiporfirin), 5 (pentakarboksiporfirin), CI (koproporfirin I), CIII (koproporfirin III), ↑↑ (značajno povišeno), ↑ (povišeno), N (uredno), V (varijabilno).
POREMEĆAJI METABOLIZMA AMINOKISELINA
Aminokiseline imaju dvojaku ulogu u organizmu: strukturna uloga (izgradnja proteina) te funkcionalna uloga (neurotransmiteri). One se dijele na esencijalne, odnosno aminokiseline koje se ne mogu sintetizirati u organizmu te moraju biti unesene u organizam putem hrane, te neesencijalne koje se mogu sintetizirati u organizmu iz različitih prekursora. Poremećaji metabolizma aminokiselina većinom su genetski uvjetovani, a tri najznačajnija poremećaja su homocistinurija i hiperhomocisteinemija, fenilketonurija te alkaptonurija.
HOMOCISTINURIJA I HIPERHOMOCISTEINEMIJA
Uvod. Homocistein nastaje kao intermedijarni produkt metabolizma metionina, esencijalne aminokiseline koja služi kao donor metilne skupine u različitim biokemijskim procesima u organizmu. Doniranjem metilne skupine metionin prelazi u homocistein, a taj je proces uvjetovan aktivnošću enzima L-metionin-S-adenoziltransferaza. Nastali homocistein može ući u dva procesa: remetilacija ili transsulfuracija. Remetilacija je proces u kojemu homocistein ponovno prima metilnu skupinu i pretvara se u metionin, pri čemu je donor metilne skupine metil-tetrahidofolat, a cijeli je proces kataliziran enzimom metilen-tetrahidrofolat reduktaza (MTHFR) uz vitamin B9 i B12 kao kofaktor. Transsulfuracija je proces koji podrazumijeva pretvorbu homocisteina u cistein što katalizira enzim cistationin-β-sintaza uz prisutnost vitamina B6 kao kofaktora. Metabolički put homocisteina ponajprije ovisi o raspoloživosti metionina u organizmu, a ako ga ima u dovoljnim količinama - homocistein dominantno ulazi u reakcije transsulfuracije, a ako ga manjka - u reakcije remetilacije. Oko 80 do 90 % homocisteina u plazmi je vezano za proteine, 5 do 10 % za cistein, dok se ostali, manji dio nalazi u slobodnom obliku koji se može lučiti iz organizma putem mokraće (količina izlučenoga cisteina putem mokraće je u fiziološkim uvjetima zanemariva).
Definicija. Homocistinurija je naziv za skupinu metaboličkih poremećaja koje obilježava nakupljanje homocisteina u plazmi (homocisteinemija) uz povećano lučenje putem urina (homocistinurija). Oni se mogu javiti kao prirođeno (nasljedno) stanje koje je posljedica poremećaja enzima uključenih u metaboličke puteve metionina i homocisteina ili kao stečeno stanje koje se vidi u nekim poremećajima.
Etiologija – nasljedna homocistinurija. Poremećaj bilo kojega od enzima koji sudjeluju u opisanim metaboličkim putevima homocisteina mogu rezultirati njegovim nakupljanjem u krvi (homocisteinemija). Dva najčešća poremećaja su deficijencija cistationin-β-sintaze te deficijencija metilen-tetrahidrofolat reduktaze, a oba se prenose autosomno-recesivnim putem nasljeđivanja.
Deficijencija cistationin-β-sintaze (klasična homocistinurija) nastaje kao posljedica mutacije CBS gena koji se nalazi na 21. kromosomu (21q22.3). Javlja se s učestalošću od 1:60 000 do 1:300 000 novorođenčadi, što ovisi o geografskoj lokaciji te metodama probira. Posljedično enzimatskom defektu dolazi do poremećaja transsulfuracije i porasta koncentracije homocisteina uz pomak metaboličkih puteva prema remetilaciji i pojačanom stvaranju metionina (hiperhomocisteinemija, hipermetioninemija).
Deficijencija metilen-tetrahidrofolat reduktaze nastaje kao posljedica mutacije MTHFR gena koji se nalazi na 1. kromosomu (1p36.3). Mutacije koje dovode do teških deficijencija ovoga enzima su rijetke (1:500 000 novorođenčadi), dok su parcijalne deficijencije puno češće i čine jedan od bitnijih rizičnih čimbenika trombofilije. Teški oblici deficijencije ovoga enzima dovode do poremećaja remetilacije i posljedične hiperhomocisteinemije uz uredne ili snižene vrijednosti metionina. MTHFR C677T polimorfizam je najčešći polimorfizam u zapadnjačkoj populaciji koji je povezan s parcijalnim deficitom ovoga enzima i posljedičnom trombofilijom.
Etiologija – stečena homocistinurija. Povišene vrijednosti homocisteina u krvi mogu se naći uslijed različitih bolesti i stanja: kronični alkoholizam, ovisnost o nikotinu, visok unos kofeina, deficijencija vitamina B skupine, šećerna bolest, psorijaza, hipotireoza, maligna oboljenja, kronična bubrežna bolest te pri uzimanju nekih lijekova (metotreksat, fenitoin, teofilin, karbamazepin).
Patogeneza. Povišene vrijednosti homocisteina mogu imati patološke učinke na koagulacijski sustav, vezivno tkivo te središnji živčani sustav.
Učinak na koagulacijski sustav. Hiperhomocisteinemija je poznat rizični čimbenik u razvoju tromboembolijskih incidenata, a svoj prokoagulantni učinak ostvaruje izazivanjem endotelne disfunkcije (direktni toksični učinak) te aktivacijom trombocita i faktora zgrušavanja.
Učinak na vezivno tkivo. Povišene vrijednosti homocisteina mogu dovesti do strukturnih promjena proteina vezivnog tkiva što ima ulogu u nastanku očnih (subluksacija leća) i koštanih poremećaja (marfanoidni aspekt). Smatra se da su promjene povezane s pojačanim vezivanjem homocisteina za cisteninske ostatke fibrilina koji je sastavni dio matriksa periosta i vezivnog tkiva očne leće.
Učinak na središnji živčani sustav. Neurološke manifestacije posljedica su agonističkoga učinka homocisteina na NDMR receptore (N-metil-D-aspartat receptor) što je povezano s pojavom konvulzija i epileptičkih napadaja uz psihološke i psihijatrijske poremećaje.
Klinička slika ovisi o etiologiji homocistinurije. Klasična homocistinurija očituje se očnim promjenama (dislokacija leće, miopija), poremećajima koštanoga sustava (marfanovski aspekt), tromboembolijskim incidentima (duboka venska tromboza, plućna tromboembolija, koronarna bolest, cerebrovaskularni incidenti) te neurološkim manifestacijama (konvulzije, epileptički napadaji, smanjene kognitivne funkcije, anksiozno-depresivno raspoloženje). Teška deficijencija metilen-tetrahidrofolat reduktaze očituje se jednako kao i klasična homocistinurija, uz iznimku pojave koštanih manifestacija. Parcijalni nedostatak metilen-tetrahidrofolat reduktaze kao i stečene homocisteinemije u pravilu su povezani s povećanim rizikom od nastanka tromboembolijskih incidenata bez ostalih navedenih manifestacija.
Dijagnoza. Osnovni korak u postavljanju dijagnoze je pobuda sumnje na homocistinuriju. Klinička slika u kojoj dominira sklonost tromboembolijskim incidentima uz ostale karakteristike (očne, koštane, neurološke) predstavlja indikaciju za određivanje ukupnoga homocisteina i metionina u plazmi. Hiperhomocisteinemija koja se ne može objasniti stečenim stanjima zahtijeva molekularnu dijagnostiku i dokazivanje mutacije CBS gena (kada je metionin povišen) ili mutacije MTHFR gena (kada je metionin uredan ili snižen). Koncentraciju homocisteina potrebno je određivati i kod osoba koje nemaju karakteristična fenotipska obilježja ovih poremećaja, a imaju osobnu ili obiteljsku anamnezu opterećenu tromboembolijskim incidentima.
Liječenje. Osnovu liječenja čini dijeta sa smanjenim unosom namirnica koje sadrže veće količine metionina, a uz to se primjenjuju vitamini B skupine (piridoksin – B6, folna kiselina – B9 te cijanokobalamin – B12), budući da potiču djelovanje preostalih enzima te time pojačavaju puteve remetilacije i transsulfuracije. Ako se ne postigne značajno sniženje vrijednosti homocisteina, u terapiju se dodaje betain koji služi kao donor metilnih skupina i potiče alternativnu remetilaciju homocisteina u metionin. Uz ove mjere svakako se preporučuju mjere prevencije tromboembolijskih incidenata (acetilsalicilna kiselina, izbjegavanje mirovanja, prestanak pušenja i uzimanja oralnih kontraceptiva i dr.).
FENILKETONURIJA
Fenilketonurija je naziv za nasljedni poremećaj metabolizma esencijalne aminokiseline fenilalanina koja se u organizam unosi putem hrane. Dio unesenoga fenilalanina koristi se za sintezu proteina, a preostali dio se uz pomoć enzima fenilalalnin hidroksilaze pretvara u tirozin uz sudjelovanje kofaktora tetrahidrobiopterina (BH4). U fenilketonuriji nalazimo mutaciju gena za fenilalanin hidroksilazu smještenoga na 12. kromosomu koja se prenosi autosomno-recesivnim putem. Posljedično manjkavosti toga enzima dolazi do akumulacije fenilalanina u organizmu te se aktiviraju alternativni katabolički putevi pri čemu nastaju različiti metaboliti – fenilketoni (fenilpiruvat, feniletilamin i drugi) koji se izlučuju putem urina po čemu je bolest i dobila ime. Osim toga, bolest može nastati i uslijed manjkavosti kofaktora tetrahidrobiopterina s obzirom na to da je i tada narušena pretvorba fenilalanina u tirozin. Bolest se očituje vrlo brzo po rođenju, a glavno kliničko obilježje je psihomotorna zaostalost razvoja djeteta uz neugodan miris urina i znoja djeteta, izazvan fenilketonima. Zbog relativno velike učestalosti ovoga poremećaja danas se u svim rodilištima radi probir na ovu bolest tako da se dijagnoza postavlja već u rodilištu. Osnova liječenja je doživotna prehrana bez fenilalanina uz dodatak tirozina. Uz pridržavanje prehrane bolesnici doživljavaju normalnu životnu dob bez intelektualnih i motoričkih ograničenja.
ALKAPTONURIJA
Alkaptonurija je rijedak nasljedni poremećaj katabolizma aminokiseline tirozina koji nastaje uslijed deficijencije oksidaze homogentizinske kiseine pri čemu izostaje njezina daljnja pretvorba u acetoacetat (prikazano na Slici 2). To uvjetuje povećanu ekskreciju homogentizinske kiseline putem urina, kao i njezinu oksidaciju pri čemu nastaje karakterističan pigment koji se nakuplja u vezivnom tkivu (ohronoza). Bolest se obično dijagnosticira u odrasloj dobi, a klinički je obilježava urin tamne do crne boje, tamna pigmentacija kože te pojava degenerativnoga artritisa uslijed nakupljanja pigmenta. Dijagnoza se potvrđuje nalazom povišene razine homogentizinske kiseline u mokraći. Specifičnoga liječenja nema, askorbinska kiselina u dozi od 1 g/dan može ublažiti odlaganje pigmenta u vezivno tkivo i zglobove pojačavajući lučenje homogentizenske kiseline urinom.
POREMEĆAJI METABOLIZMA ŽELJEZA
Metabolizam željeza. Željezo je bitan oligoelement u ljudskom organizmu koji ulazi u sastav različitih proteina. U organizmu dolazi u dva oblika, kao dvovalentno ili trovalentno željezo, a razmjenom elektrona sudjeluje u stvaranju slobodnih kisikovih radikala te se zbog toga u stanicama i krvi prenosi vezan za proteine. Dnevna apsorpcija željeza iz hrane putem probavnoga sustava iznosi oko 1 do 2 mg i ona je jednaka prosječnom dnevnom gubitku koji se ostvaruje deskvamacijom epitelnih stanica probavnoga sustava i epidermisa, gdje se željezo nalazi uskladišteno u obliku feritina, ili menstruacijskim krvarenjem kod žena. Apsorpcija željeza odvija se preko enterocita u duodenumu i proksimalnom jejunumu, a da bi se željezo moglo apsorbirati, ono mora biti u dvovalentnom obliku, tako da se trovalentno željezo iz hrane najprije mora reducirati unutar probavne cijevi. Nakon ulaska u enterocite željezo se može vezati za apoferitin pri čemu nastaje feritin, skladišni oblik željeza, ili se putem feroportina izbacuje u cirkulaciju. U cirkulaciji se željezo može nalaziti u svome slobodnom obliku (manji, zanemariv dio), dok se veći dio željeza veže na apotransferin i formira transferin koji prenosi željezo do ciljnih stanica. Vezanje željeza za transferin omogućava ceruloplazmin. On posreduje otpuštanje željeza iz feroportina, tj. vrši njegovu oksidaciju iz dvovalentnoga u trovalentni oblik kako bi se mogao vezati na apotransferin te u njegovom nedostatku željezo neće biti otpušteno s feroportina. Ulazak željeza koji se nalazi u molekuli transferina u ciljne stanice omogućuju transferinski receptori 1 i 2 (TRF1, TRF2). Ovu interakciju transferina i transferinskih receptora regulira HFE protein, produkt HFE gena. Novija istraživanja ukazuju na to da osim transferina postoje i drugi prijenosnici željeza, kao što je protein H. Stanice mogu preuzeti i željezo iz hemoglobina koji se u svom slobodnom obliku, izvan eritrocita, normalno nalazi u malim količinama u krvi, ali se one mogu povećati u uvjetima hemolize. Da bi se spriječio njegov gubitak putem bubrega, hemoglobin se u krvi veže za haptoglobin koji ga potom prenosi do retikuloendotelnih stanica jetre, slezene i koštane srži. Hepatociti i stanice retikuloendotelnoga sustava (makrofagi, monociti) predstavljaju glavna mjesta skladištenja željeza u kojima se on pohranjuje u obliku feritina i hemosiderina na koji se željezo veže kada se popuni sva zaliha apoferitina. 80 % željeza u organizmu koristi se za sintezu hemoglobina, a ostatak za sintezu drugih proteina i enzima. Budući da organizam nema razvijen ekskrecijski put za željezo, u homeostazi ovoga oligoelementa ključnu ulogu ima dobro regulirana apsorpcija koja treba odgovarati dnevnim gubicima željeza. U slučaju nedostatka željeza u organizmu najprije dolazi do njegova oslobađanja iz skladišnih oblika (feritin, transferin) koji se potom predaje apotransferinu u cirkulaciji, dok se u obrnutoj situaciji većina željeza predaje u stanice pri čemu se formiraju feritin i hemosiderin. Jedan od glavnih regulatora razine željeza u krvi je hepcidin, protein koji se sintetizira u jetri u stanjima povećane količine željeza u krvi, što detektiraju tzv. senzori za željezo koji se nalaze na membranama hepatocita (npr. hemojuvelin). On djeluje tako da smanjuje ekspresiju feroportina i onemogućava izlazak željeza iz enterocita i drugih stanica u cirkulaciju. Na pojačano stvaranje hepcidina utječu i druga stanja kao što su infekcije, dok se smanjeno stvaranje hepcidina bilježi kod sniženih vrijednosti željeza u krvi, anemije i hipoksije (Slika 1).
Poremećaji metabolizma željeza mogu se podijeliti na one koji dovode do povećanoga sadržaja i koji dovode do smanjenoga sadržaja željeza u organizmu. Poremećaji koji dovode do povećanoga sadržaja željeza mogu biti nasljedni (hemokromatoza, aceruloplazminemija) te stečeni (masivne transfuzije krvi, terapijska primjena željeza). Poremećaji koji dovode do smanjenoga sadržaja željeza u organizmu također mogu biti nasljedni (IRDA, engl. iron-refractory iron-deficiency anemia) i stečeni (sideropenična anemija, anemija kronične bolesti). U nastavku će biti opisani osnovni poremećaji metabolizma željeza, a to su hemokromatoza (nasljedna i stečena) te aceruloplazminemija. Sideropenija i sideropenična anemija opisani su u sklopu hematologije.
NASLJEDNA HEMOKROMATOZA
Definicija. Nasljedna ili hereditarna hemokromatoza (HH) je naziv za skupinu genetski uvjetovanih poremećaja metabolizma željeza koji su karakterizirani pojačanom apsorpcijom željeza iz probavne cijevi i posljedičnim nakupljanjem u različitim tkivima i organima gdje dovodi do oštećenja i poremećaja funkcije. Ukupno gledano, radi se o jednome od najčešćih genetskih metaboličkih poremećaja u općoj populaciji. Bolest se češće javlja kod muškaraca u odnosu na žene (6:1).
Etiopatogeneza. Nasljedna hemokromatoza može biti uzrokovana mutacijom jednoga od pet gena čiji proteinski produkti sudjeluju u metabolizmu željeza te se prema tome razlikuje pet tipova ovoga poremećaja (Tablica 8.22.). Zajednička osnova svih poremećaja je narušena regulacija apsorpcije željeza iz probavne cijevi pri čemu se ono apsorbira u povećanim količinama s posljedičnim nakupljanjem u krvi i drugim tkivima i organima. Tijekom jedne godine dođe do akumulacije 0,5 do 1,0 g željeza što za 20 do 30 godina dovede do nakupljanja višestruko veće količine nego je to normalno (5 - 6 g). Povećana intracelularna koncentracija željeza dovodi do direktnoga staničnog oštećenja uvjetovanoga povećanim stvaranjem kisikovih radikala što vodi u kroničnu upalu, a kasnije i fibrozu s ireverzibilnim gubitkom funkcije.
|
Tablica 8.22. Klinički oblici nasljedne hemokromatoze
|
|
|
Tip I
|
Tip IIa
|
Tip IIb
|
Tip III
|
Tip IV
|
|
Gen
|
HFE (6p21.3)
|
HJV (1q21)
|
HAMP (19q13.1)
|
TRF2 (7q22)
|
SLC40A1 (2q32)
|
|
Produkt
|
HFE protein
|
Hemojuvelin
|
Hepcidin
|
TRF2 receptor
|
Feroportin
|
|
Mutacija
|
C282Y, H63D
|
G320V
|
93delG
|
Y250X
|
V162del
|
|
Prijenos
|
AR
|
AR
|
AR
|
AR
|
AD
|
|
Pojava
|
Srednja i starija dob
|
Mlađa dob
|
Mlađa dob
|
Srednja dob
|
Srednja i starija dob
|
Više od 85 % slučajeva hemokromatoze čine homozigoti za C282Y mutaciju, a smatra se da je učestalost homozigota za navedenu mutaciju oko 0,5 % u općoj populaciji. Više od 30 % takvih osoba neće imati klinički manifestnu bolest (klinički i laboratorijski pokazatelji) što ukazuje na različitost u penetraciji gena i ekspresije bolesti.
Patologija. Glavno patološko obilježje hemokromatoze je pojačano nakupljanje željeza u obliku feritina i hemosiderina u brojnim organima i tkivima, što se može dokazati bojanjem berlinskim modrilom. Najčešće zahvaćeni organi su jetra, gušterača, srce, endokrine žlijezde (hipofiza, štitnjača, gonade) te koža i zglobovi. Početne promjene uključuju upalni proces nakon kojega slijedi nekroza i na kraju fibroza.
Klinička slika. Bolesnici su kroz dug period bez simptoma i tada se mogu dijagnosticirati samo na osnovu patoloških laboratorijskih nalaza. U kasnijem tijeku pojavljuju se nespecifični simptomi kao što su umor, slabost, gubitak na tjelesnoj težini i opća apatija te specifični simptomi koji ovise o zahvaćenom organu (Tablica 8.23.).
|
Tablica 8.23. Kliničke prezentacije hemokromatoze
|
|
Jetra
|
Koža i zglobovi
|
Endokrine žlijezde
|
Ostalo
|
|
Asimptomatski patološki laboratorijski nalazi
Hepatosplenomegalija
Ciroza i zatajenje jetre
|
Artropatije
Brončana boja kože
|
Hipogonodizam
Hipotireoidizam
Diabetes mellitus
Hipopituitarizam
|
Kardiomiopatija
Poremećaji srčanoga ritma
Srčano popuštanje
|
Dijagnoza se postavlja na osnovi kliničke sumnje, biokemijskih laboratorijskih parametara, patohistološkoga nalaza biopsije jetre te genetske analize.
Biokemijski laboratorijski parametri koje određujemo kod sumnje na hemokromatozu su vrijednosti serumskoga željeza, serumskoga feritina te zasićenje transferina koji su povišeni kod bolesnika s hemokromatozom (Tablica 8.24.). Budući da sva tri parametra mogu biti povišena i uslijed brojnih drugih stanja i bolesti, nisu dovoljno specifični niti osjetljivi za postavljanje dijagnoze hemokromatoze (najosjetljiviji je serumski feritin jer je za njegovo povišenje potrebna značajno veća količina tjelesnoga željeza). Druge bolesti i stanja koje mogu dovesti do povišenja tih vrijednosti su upalne bolesti, maligne bolesti, alkoholizam, trudnoća te prehrana. Da bi se izbjegao utjecaj hrane na razinu serumskoga željeza, preporuča se uzorkovanje krvi za tu pretragu ujutro natašte.
Biopsija jetre donedavno je predstavljala zlatni standard u postavljanju dijagnoze hemokromatoze kod bolesnika koji su imali patološke biokemijske markere, no širokom uporabom molekularnih metoda i otkrivanjem specifičnih mutacija, ona je danas od manjega značaja. Patohistološki se može dokazati različit stupanj jetrene lezije (od masne promjene do ciroze), a bojanjem berlinskim modrilom moguće je i dokazati depozite željeza u hepatocitima. Ključna analiza kod ovih bolesnika je mjerenje koncentracije željeza u jetrenom tkivu, tzv. indeks jetrenoga željeza (HII, engl. hepatic iron indeks) koji se izračunava dijeljenjem količine željeza u jetri (µmol/g suhe jetre) s bolesnikovim godinama, a dobivena vrijednost veća od 1.9 indikativna je za hemokromatozu. Uvođenjem genetske dijagnostike pokazano je da je kod mnogih bolesnika s homozigotnim mutacijama za hemokromatozu taj indeks manji od 1,9 te se stoga i on pokazao nepouzdanim. S obzirom na izneseno, biopsija jetre nije nužna kod svih bolesnika ako su dostupna genetska testiranja. U slučaju genetski potvrđene hemokromatoze, biopsija jetre izvodi se samo s ciljem dokazivanja ciroze, a na to ukazuju vrijednost feritina > 1000 ng/ml te povišene jetrene transaminaze.
Genetsko testiranje nasljedne hemokromatoze danas predstavlja pretragu izbora kod sumnje na ovu bolest. Na taj se način otkrivaju i dokazuju specifične mutacije za ovu bolest, a najčešće se radi o homozigotima C282Y/ C282Y (najčešće mutacije prikazane su u prethodnoj Tablici 8.22.). Indicirano je za sve osobe sa sumnjom na hemokromatozu (klinička slika, patološki biokemijski pokazatelji) te u probiru članova obitelji bolesnika s dijagnosticiranom hemokromatozom.
|
Tablica 8.24. Biokemijski pokazatelji hemkokromatoze
|
|
Serumsko željezo
> 35 µmol/L
|
Zasićenje transferina
> 50 % žene
> 60 % muškarci
|
Serumski feritin
> 300 ng/ml muškarci i žene u menopauzi
> 200 ng/ml žene u premenopauzi
|
Liječenje hemokromatoze u prvom redu uključuje izbjegavanje hrane bogate željezom uz primjenu terapijskih venepunkcija, dok je primjena kelatora željeza rezervirana samo za one bolesnike koji ne toleriraju venepunkcije.
Terapijska venepunkcija indicirana je kod svih simptomatskih bolesnika te kod bolesnika čije su vrijednosti feritina veće od 1000 ng/ml. Početno se rade tjedne venepunkcije kojima se ukloni 250 - 500 ml krvi, a s time se izgubi oko 250 do 500 mg željeza. Praćenje ovih bolesnika sastoji se od određivanja vrijednosti hemoglobina i hematokrita prije svake nove venepunkcije te određivanja feritina i zasićenosti transferina svaka tri mjeseca. Određivanjem hemoglobina i hematokrita izbjegava se razvoj sideropenične anemije, dok su vrijednosti feritina i zasićenosti transferina bitne u procjeni nastavka terapije. Kada vrijednosti feritina padnu ispod 100 ng/ml i zasićenje transferinom ispod 50 % , venepunkcije se izvode jednom u tri mjeseca. Takav način liječenja dokazano poboljšava simptome bolesti, osobito ako se započne na vrijeme i nisu se razvile ireverzibilne promjene na pojedinim organima.
Kelatori željeza su lijekovi koji se primjenjuju kod ovih bolesnika s ciljem odstranjenja željeza u suvišku, a rezervirani su za bolesnike koji ne toleriraju venepunkcije ili su razvili anemiju. Na raspolaganju je pripravak za intravensku primjenu (deferoksamin) te dva pripravka za peoralnu primjenu (deferasiroks i deferiprone). Deferoksamin se primjenjuje u dozi od 20 - 49 mg/kg/dan te se infundira tijekom 24 sata i tijekom jedne takve terapije može se mobilizirati do 30 mg željeza. Zbog toksičnih učinaka (oko, uho), bolova pri apliciranju te potrebnoga vremena, ovaj lijek se rijetko primjenjuje. Deferasiroks se primjenjuje u dozi od 20 mg/dan u jednoj dozi, dok se deferipron primjenjuje u dozi od 25 mg/kg tri puta na dan.
Prognoza. Bolesnicima kojima se bolest dijagnosticira u presimptomatskoj fazi u kojoj nisu nastupile ireverzibilne promjene na pojedinim organima uz pridržavanje terapije životni je vijek jednake očekivane dužine kao i općoj populaciji. Nakon nastupa ireverzibilnih promjena (ciroza, kardiomiopatija, šećerna bolest) bolesnici su ugroženi zbog komplikacije koje nose te bolesti. Ciroza jetre kod ovih bolesnika često se komplicira razvojem hepatocelularnoga karcinoma pa je tu skupinu bolesnika potrebno češće promatrati.
STEČENA HEMOKROMATOZA
Stečena hemokromatoza odnosi se na stanje povećanoga nakupljanja željeza u organizmu koje nije uzrokovano genetskim poremećajima, nego različitim stečenim bolestima i stanjima.
Najčešće bolesti i stanja koje dovode do stečene hemokromatoze su anemije (neučinkovita eritropeza, često liječenje transfuzijama), alkoholna ciroza jetre, nealkoholni steatohepatitis, kronični virusni hepatitis, defekti transporta željeza, porfirija kutanea tarda, stanje nakon portokavalnoga šanta, uzimanje velikih količina željeza prehranom te terapijska primjena željeza i dekstrana.
Dijagnostika i liječenje sekundarne hemokromatoze provodi se u sklopu liječenja osnovnih poremećaja. Za razliku od nasljedne hemokromatoze, vrijednosti biokemijskih parametara (serumsko željezo, serumski feritin, zasićenje transferina) nisu toliko povišene.
ACERULOPLAZMINEMIJA
Definicija. Aceruloplazminemija je rijetka autosomno-recesivna nasljedna bolest koju karakterizira poremećaj stvaranja ceruloplazmina, proteina koji ima ulogu u transportu bakra i željeza.
Etiopatogeneza. Posljedično mutaciji gena za ceruloplazmin nastaje abnormalan i afunkcionalni protein koji ne može vezati željezo na sebe te na taj način smanjuje prijenos željeza iz tkiva. Ceruloplazmin posreduje otpuštanje željeza iz feroportina, tj. vrši njegovu oksidaciju iz dvovalentnoga u trovalentni oblik kako bi se mogao vezati na apotransferin. U nedostatku ceruloplazmina feroportin neće otpustiti željezo u cirkulaciju te će se on pojačano nakupljati u stanicama.
Klinička slika. Bolest se obično prezentira od drugoga do šestog desetljeća života, a karakterizirana je degeneracijom mrežnice, šećernom bolesti i neurološkim simptomima koji uključuju distoniju, tremor, dizartriju, ataksiju i blefarospazam.
Dijagnoza se postavlja na temelju kliničke slike, slikovnih metoda (magnetna rezonanca mozga) te uz karakteristične patološke laboratorijske nalaze (snižen ceruloplazmin, povišen feritin, snižena zasićenost transferinom, snižen bakar). Genetske analize mutacija gena za ceruloplazmin nisu komercijalne i vrše se trenutno samo u istraživačke svrhe.
Liječenje se provodi primjenom kelatora željeza (kako je opisano kod nasljedne hemokromatoze), a dobro se pokazala i primjena svježe smrznute plazme i antioksidanasa (vitamin E).
POREMEĆAJI METABOLIZMA BAKRA
Metabolizam bakra. Bakar je esencijalni element koji u organizmu ima višestruke uloge, s obzirom na to da je sastavni dio različitih enzima i enzimatskih sustava. Molekularni mehanizmi homeostaze genetski su uvjetovani i u njima sudjeluju različiti geni i njihovi proteinski produkti koji ponajprije služe kao membranski prijenosnici bakra. U normalnim okolnostima hranom se u organizam unese oko 1 do 3 mg bakra, od kojega se oko 50 % apsorbira iz gornjega dijela probavne cijevi (želudac, početni dio tankoga crijeva) što je uvjetovano produktom ATP7A gena, transmembranskom ATP-azom koja pomaže prijenos bakra u cirkulaciju gdje se veže na albumine i kao takav dolazi do jetre te uz pomoć istoga proteina ulazi u hepatocite. Unutar hepatocita bakar ulazi u jedan od metaboličkih puteva: 1) sudjeluje u izgradnji ceruloplazmina (transportni protein za bakar i željezo), 2) sudjeluje u izgradnji specifičnih enzima te 3) višak bakra se izlučuje u žuč te njome odlazi u tanko crijevo. Ovi procesi posredovani su sličnom transmembranskom ATP-azom, produktom ATP7B gena. Bakar se u cirkulaciji prenosi vezan na albumine i ceruloplazmin te je kao takav dostupan drugim stanicama i tkivima u kojima ima određene fiziološke uloge.
Poremećaji metabolizma bakra. Dva najčešća poremećaja metabolizma bakra nastaju kao posljedica mutacije ATP7A gena (Menkesova bolest) koja se očituje poremećajem apsorpcije bakra iz probavne cijevi i njegovom posljedičnom deficijencijom u organizmu te kao posljedica mutacije ATP7B gena (Wilsonova bolest) koja se očituje poremećenim izlučivanjem bakra iz organizma i posljedičnim nakupljanjem u raznim organima i tkivima.
MENKESOVA BOLEST
Menkesova bolest je autosomno-recesivni poremećaj koji nastaje uslijed mutacije ATP7A gena koji se nalazi na X-kromosomu (X-vezano nasljeđivanje). Posljedično genetskom defektu dolazi do poremećaja apsorpcije bakra iz probavnoga sustava, zbog čega se javlja njegova deficijencija. Učestalost bolesti je 1:100 000 do 1:250 000, očituje se vrlo brzo po rođenju, a djeca ne dožive više od tri godine.
WILSONOVA BOLEST
Definicija. Wilsonova bolest (progresivna hepatolentikularna degeneracija) naziv je za poremećaj metabolizma bakra pri kojem dolazi do njegovoga patološkog nakupljanja najprije u jetri, a zatim i drugim organima, ponajprije mozgu (bazalni gangliji), rožnici i bubrezima. Učestalost Wilsonove bolesti iznosi oko 1:30 000 novorođenčadi.
Etiopatogeneza. Radi se o autosomno-recesivnoj nasljednoj bolesti kod koje nalazimo mutaciju ATP7B gena s posljedičnim poremećajem transmembranske ATP-aze koja dovodi do smanjenoga lučenja bakra iz hepatocita u žuč i feces (jedini eliminacijski mehanizam bakra iz tijela), kao i poremećaja sinteze ceruloplazmina. ATP7B gen nalazi se na 13. kromosomu i do danas je poznato više od 500 različitih mutacija toga gena, a u više od 50 % slučajeva radi se o H1069Q mutaciji. Bakar se najprije nakuplja unutar hepatocita što dovodi do oksidativnoga oštećenja koje vodi u upalu i fibrozu. Ako se bolest ne prepozna i ne liječi, dolazi do nakupljanja bakra i u drugim tkivima i organima, posebno unutar bazalnih ganglija u mozgu, rožnici (Kayser-Fleischerov prsten) te bubrezima (tubularna degeneracija). Posljedično poremećenoj sintezi ceruloplazmina, njegove vrijednosti u serumu su snižene.
Patologija. Glavne i najranije promjene vide se u tkivu jetre, a s obzirom da je ona i najdostupnija za uzorkovanje, patohistološki nalaz jetre ključan je u potvrdi dijagnoze Wilsonove bolesti. Promjene ovise i stadiju bolesti i kreću se od masne promjene, nekroze i upale pa sve do fibroze i razvoja mikronodularne ciroze uz karakteristični nalaz povećane koncentracije bakra u suhom tkivu jetre (> 250 µg/g suhoga tkiva jetre).
Klinička slika. Bolest se obično klinički prezentira do 40. godine života pri čemu u djetinjstvu i mlađoj dobi dominiraju simptomi i znaci zahvaćenosti jetre, dok se neurološki simptomi javljaju kasnije, iako ima opisanih slučajeva i pojavnosti simptoma i znakova bolesti nakon petoga desetljeća života. Prema najčešće zahvaćenim tkivima i organima, simptome možemo podijeliti u nekoliko skupina: jetrene manifestacije, neurološke manifestacije, psihijatrijske manfestacije te ostale manifestacije.
Jetrene manifestacije uključuju širok dijapazon kliničkih poremećaja: od asimptomatskih poremećaja laboratorijskih nalaza jetrenih transaminaza, preko kliničke slike akutnoga i kroničnog hepatitisa pa do brzoprogresivnoga jetrenog zatajenja ili razvoja dekompenzirane ciroze jetre. Simptomi i znakovi vezani uz jetru (hepatomegalija, ikterus, bol pod desnim rebrenim lukom, razvoj protralne hipertenzije i dr.) najraniji su znakovi Wilsonove bolesti i obično se počinju razvijati u djetinjstvu, i ako se ne prepoznaju i ne liječe postupno se pogoršavaju kako bolest napreduje prema cirozi.
Neurološke manifestacije javljaju se kasnije, obično u mlađoj odrasloj životnoj dobi, a primarno su posljedica poremećaja bazalnih ganglija: poremećaji kretnji (tremor, distonija, rigidnost, koreiformni pokreti), dizartrija, disfagija, česte su i migrenozne glavobolje, nesanica i pojava epileptičkih napadaja.
Psihijatrijske manifestacije povezane su s neurološkim manifestacijama, najčešće se očituju depresivnim raspoloženjem, a mogući su i psihotični poremećaji te različite promjene osobnosti.
Kayser-Fleischerov prsten je patognomonični znak za ovu bolest, a nastaje kao posljedica odlaganja bakra u obliku kružnice u rožnici. Opisuje se kao zelenkasto-smećkast anularni depozit uz vanjski rub rožnice koja se uglavnom može vidjeti već golim okom ili se uočava tijekom oftalmološkoga pregleda. Javlja se kod 70 % bolesnika s dominantnim jetrenim manifestacijama i kod više od 95 % bolesnika s neurološkim manifestacijama.
Ostale manifestacije rjeđe se javljaju, a povezane su s odlaganjem bakra u druga tkiva i organe. Najčešće su to bubrezi s posljedičnim razvojem tubularnih poremećaja (aminoacidurija, nefrolitijaza), koštano-zglobni sustav (artritis, osteoporoza), srce (kardiomegalija, poremećaji ritma i provođenja), endokrinološki poremećaji i drugi.
Dijagnoza se postavlja na temelju nalaza biokemijskih poremećaja, patohistološkoga nalaza jetre te molekularnim metodama dokazivanja genetskoga poremećaja. Ključni korak u postavljanju dijagnoze je pomisao na samu bolest, a sumnju na nju trebaju pobuditi različiti kliničko-laboratorijski pokazatelji (Tablica 8.25.).
|
Tablica 8.25. Kliničko-laboratorijski pokazatelji Wilsonove bolesti
|
- Jetrena bolest nejasne etiologije u adolescentskoj dobi
- Neuropsihijatrijski poremećaji nejasne etiologije u ranoj odrasloj dobi
- Povišenje jetrenih enzima s obično sniženom alkalnom fosfatazom
- Omjer AST:ALT veći od 2.2
- Omjer alkalna fosfataza (units/L):ukupni bilirubin (mg/dL) manji od 4
- Coombs negativna hemolitička anemija
|
Biokemijski poremećaji. Kod sumnje na Wilsonovu bolest ključne laboratorijske pretrage su određivanje serumskoga ceruloplazmina i bakra te određivanje bakra u 24-satnom urinu.
Normalne vrijednosti serumskoga ceruloplazmina iznose od 25 do 35 mg/dL, vrijednosti od 5 do 10 mg/dL izazivaju temeljitu sumnju na Wilsonovu bolest, dok vrijednosti manje od 5 mg/dL gotovo sa sigurnošću ukazuju na tu bolest. Nasuprot tome, vrijednosti ceruloplazmina veće od 30 mg/dL isključuju mogućnost Wilsonove bolesti. Pri interpretaciji nalaza treba voditi računa o sljedećim napomenama: kod 15 % bolesnika s Wilsonovom bolesti vrijednosti ceruloplazmina mogu biti uredne ili na donjoj granici; osobe koje su heterozigoti za ovu bolest mogu imati snižene vrijednosti ceruloplazmina (< 20 mg/dL); sniženim vrijednostima ceruloplazmina pogoduje pothranjenost, a povišenima oralni kontraceptivi, steroidi i estrogeni.
Vrijednosti bakra u serumu su snižene, iako je ukupna količina bakra u tijelu povišena, što je razlog smanjenom stvaranju ceruloplazmina. Kada kod akutnoga i fulminantnog hepatitisa dolazi do nekroze hepatocita može doći do otpuštanja bakra u cirkulaciju s posljedičnim povišenim vrijednostima.
Normalne vrijednosti bakra u 24-satnom urinu iznose 25 - 50 µg, a vrijednosti veće od 100 ukazuju na Wilsonovu bolest. Treba imati na umu da povišene vrijednosti (kupriurija) mogu nastati i zbog drugih razloga, kao što je kontaminacija bakrom iz posuda u koje se prikuplja urin ili kronična bolest jetre koja traje dulje od jedne godine te povišene vrijednosti možemo naći kod heterozigota. Specifičniji nalaz daje određivanje vrijednosti bakra nevezanoga na ceruloplazmin, čije normalne vrijednosti iznose 50 do 50 mcg/L.
Biopsija jetre jedan je od ključnih koraka u dijagnostici Wilsonove bolesti. Pored opisanih patomorfoloških promjena, u uzorku se određuje količina bakra, a kod bolesnika s ovom bolesti nalazimo više od 250 µg/g suhoga tkiva jetre. Povećanu količinu bakra u hepatocitima imaju i bolesnici s kroničnim kolestatskim bolestima.
Genetska dijagnostika, odnosno molekularna dijagnostika mutacija ATP7B gena moguća je, iako ima ograničenu ulogu, pošto je do danas poznato više od 500 mutacija.
Ostale dijagnostičke metode uključuju magnetsku rezonancu mozga za dokaz nakupina bakra u bazalnim ganglijima te biopsiju bubrega u nejasnim slučajevima poremećaja bubrežne funkcije.
Liječenje. Osnovni korak u liječenju Wilsonove bolesti je dijetni režim te primjena kelatora bakra. Dijetni režim podrazumijeva izbjegavanje hrane koja sadrži veće količine bakra (orašasto voće, morski plodovi, čokolada, mahunarke i jetrica). U kliničkoj praksi primjenjuju se sljedeći kelatori: penicilamin, trientin te pripravci cinka. Liječenje treba započeti što ranije i treba se provoditi doživotno.
Penicilamin predstavlja lijek izbora u liječenju Wilsonove bolesti koji veže absorbirani bakar i povećava njegovo izlučivanje putem urina. Daje se u dozi od 0,75 - 2 g/dan, podijeljeno u dvije doze koje se uzimaju jedan sat prije jela ili dva sata nakon jela. Lijek vrlo brzo dovodi do sniženja bakra u serumu i preporuča se započeti terapiju što prije, još u asimptomatskoj fazi, kako bi se izbjegla trajna oštećenja organa. Glavna ograničenja primjene ovoga lijeka proizlaze iz mogućih nuspojava: gastrointestinalne smetnje, autoimune reakcije (engl. lupus like syndrome), supresija koštane srži (leukopenija), nefrotoksičnost i kožne promjene (pemfigoidne promjene). Zbog toga takve bolesnike treba češće pratiti i kontrolirati. Također, lijek djeluje kao antimetabolit vitamina B 6 pa se preporuča istovremena primjena piridoksina u dozi od 50 mg/tjedan.
Trientin ima sličan mehanizam djelovanja kao i penicilamin uz značajno manje nuspojava. Nekada se poglavito koristio kod bolesnika koji nisu mogli uzimati penicilamin, a danas se sve više koristi i kao prva linija u liječenju Wilsonove bolesti. Lijek se uzima u dozi od 250 do 500 mg tri puta na dan.
Liječenje penicilaminom ili trientinom treba se provoditi sve dok vrijednosti serumskoga bakra nevezanog za ceruloplazmin ne dođu u referentne vrijednosti (50 - 150 mcg/L) kada se oni postupno smanjuju na najmanju moguću dozu kojom se postiže kontrola bolesti ili po mogućnosti ukidaju uz nastavak liječenja cinkom.
Preparati cinka (cink-acetat, cink-glukonat) smanjuju intestinalnu apsorpciju bakra i tako potiču lučenje bakra putem stolice, a primjenjuju se u dozi od 50 mg tri puta na dan. Glavne indikacije su primjena kod asimptomatskih bolesnika te kao terapija održavanja kod bolesnika koji su liječeni navedenim kelatorima i kod kojih je postignuta stabilizacija bolesti (kontrola biokemijskih parametara).
Ostale terapijske napomene. U liječenju se primjenjuju i sve druge standardne terapijske opcije ako dođe do razvoja ciroze jetre, neuroloških i drugih manifestacija. Preporuča se i suplementacija vitamina E zbog anioksidativnoga učinka (hepatoprotekcija). Trasplantacija jetre indicirana je pri fulminantnom hepatitisu.
Prognoza. Rano prepoznavanje bolesti i brzi početak liječenja značajno poboljšavaju konačne ishode bolesti. Određen stupanj poboljšanja može se postići kod bolesnika s razvijenom jetrenom ili neurološkom bolesti, iako su neurološke promjene u pravilu ireverzibilne.
POREMEĆAJI KOŠTANOGA METABOLIZMA
Uvod. Kako bi se razumjeli osnovni patofiziološki procesi i poremećaji koštanoga metabolizma, potrebno je poznavati osnove funkcije i interakcije koštanih stanica, kao i osnove utjecaja minerala i hormona na koštani metabolizam. Koštano tkivo sastoji se od koštanih stanica i koštanoga matriksa koji podrazumijeva organski (kolagen, proteoglikani) i anorganski dio (kalcij, fosfati). Koštano tkivo je metabolički aktivno tkivo koje karakterizira koštana pregradnja sačinjena od povezanoga slijeda koštane razgradnje i izgradnje. Koštana pregradnja započinje aktivacijom osteoklasta koju slijede osteoblasti koji zajedno tvore organizacijsku i funkcionalnu jedinicu koja se naziva temeljno višestanično ustrojstvo (BMU, engl. basic multicellular unit). Na taj se način prosječno izmjeni 8 do 10 % ukupnoga koštanog tkiva tijekom jedne godine.
FUNKCIJA I INTERAKCIJE KOŠTANIH STANICA
Osteoblasti su koštane stanice koje izgrađuju kost, odnosno sintetiziraju koštani matriks te reguliraju njegovu mineralizaciju. Oni nastaju od mezenhimalnih matičnih stanica, a u tome procesu najbitniju ulogu ima transkripcijski faktor Runx2 koji kontrolira ekspresiju različitih molekula bitnih za sazrijevanje osteoblasta. Kod transgeničkih miševa s potpunom deficijencijom Runx2 izostaje razvoj koštanoga tkiva (imaju potpun nedostatak osteoblasta), dok kod djelomične deficijencije nalazimo odgođeno formiranje kosti. Runx2 ekspresija regulirana je od strane koštanih morfogenetskih proteina (BMP, engl. bone morphogenetic proteins) te čimbenika rasta Ihh (engl. Indian hedgehog). Na površini osteoblasta nalazimo brojne receptore za različite faktore rasta (transfomirajući faktor rasta, faktor rasta trombocita, inzulinu-sličan faktor rasta, faktor rasta fibroblasta) i hormone (paratireoidni hormon, esterogen, glukokortikoidi, vitamin D) koji utječu na njihovu funkciju i aktivnost. Osim toga, osteoblasti utječu na aktivnost osteoklasta i to putem RANK liganda (RANKL, od engl. receptor activator of nuclear factor kB ligand) koji se pojavljuje na površini osteoblasta i potiče aktivaciju osteoklasta te putem osteoprotegerina (OPG) koji se luči iz osteoblasta te ometa sazrijevanje/aktivaciju osteoklasta (opisano kasnije).
Osteociti nastaju od osteoblasta nakon što prestane njihova aktivnost i okruži ih novonastala kost, a međusobno su povezani citoplazmatskim nastavcima koji prolaze kroz koštane kanaliće. Osteociti reagiraju na mehaničko opterećenje kosti (koštani mehanoreceptori) te različitim signalnim putevima taj signal prenose na osteoblaste i osteoklaste i na taj način potiču koštanu pregradnju.
Osteoklasti su koštane stanice koje razgrađuju kost, a radi se o multinuklearnim stanicama koje nastaju spajanjem prekursora makrofaga u koštanoj srži, što je potaknuto djelovanjem faktora koji stimulira kolonije makrofaga (M-CSF, engl. macrophage colony stimulating factor). Daljnje sazrijevanje i aktivacija osteoklasta potaknuti su djelovanjem osteoblasta i drugih citokina. Osteoklasti na svojoj površini sadrže RANK receptore za koje se mogu vezati RANK ligandi koji potiču sazrijevanje i aktivaciju osteoklasta. Ovu staničnu interakciju mogu ometati OPG molekule koje stvaraju osteoblasti, a koje mogu oponašati RANK receptore i vezati se na RANK ligande te na taj način onemogućiti djelovanje RANK liganda na osteoklaste. Osim osteoblasta, na funkciju osteoklasta utječu razni citokini kao što su TNF-α, interferon-γ, IL-1, IL-6 te IL-11. Paratireoidni hormon i vitamin D neizravno utječu na funkciju osteoklasta preko osteoblasta, dok kalcitonin izravno djeluje vežući se za receptore na površini osteoklasta i inhibira njihovu aktivnost.
HORMONI I MINERALI UKLJUČENI U KOŠTANI METABOLIZAM
Kalcij i fosfati predstavljaju osnovne minerale koji ulaze u sastav koštanoga matriksa koji osim toga imaju i druge bitne fiziološke uloge u organizmu. Fosfati sudjeluju u procesima dobivanja energije u aerobnim uvjetima, dok kalcij sudjeluje u neuromišićnoj transmisiji, koagulacijskom procesu te mišićnoj kontrakciji. Kalcij u plazmi dolazi u dva oblika: polovica dolazi u slobodnom obliku (ionizirani, biološki aktivni), dok je druga polovica u vezanom obliku (80 % vezano za albumine, 20 % vezano za druge anione). Održavanje homeostaze kalcija i fosfata u krvi usko je povezano s učinkom paratireoidnoga hormona i vitamina D.
Paratireoidni hormon (PTH) je glavni fiziološki regulator koncentracije kalcija i fosfata u krvi. On se sintetizira u stanicama paratireoidne žlijezde, a poticaj za njegovo izlučivanje je snižena razina kalcija u krvi. Ima nekoliko značajnih učinaka: 1) potiče mobilizaciju kalcija iz kosti (aktivacija osteoklasta); 2) smanjuje gubitak kalcija putem bubrega povećavajući tubularnu reapsorpciju kalcija; 3) neizravno potiče apsorpciju kalcija iz crijeva stimulirajući nastanak aktivnoga oblika vitamina D (kalcitriol) te 4) potiče izlučivanje fosfata putem bubrega. Konačni rezultat djelovanja PTH-a je povećanje razine kalcija uz smanjenje razine fosfata. Povećane razine kalcija registriraju posebni receptori na površini stanica paratireoidne žlijezde čime se koči lučenje PTH-a (mehanizam negativne povratne sprege).
Vitamin D ima sličan učinak kao i PTH. U organizmu vitamin D djeluje kao hormon, a dolazi u dva oblika: ergokalciferol (D2) koji se u organizam unosi putem hrane te kolekalciferol (D3) koji nastaje u koži iz 7-dehidrokolesterola pod utjecajem UV zraka. Kolekalciferol se dalje u jetri pretvara u 25-hidroksi vitamin D3 (kalcifediol), a on u bubrezima u 1,25-dihidroksikolekalciferol (kalcitriol). Ovaj završi korak pretvorbe kalcifediola u kalcitriol u najvećoj mjeri potiče PTH. Glavni učinci kalcitriola su pojačana apsorpcija kalcija iz crijeva, pojačana mobilizacija kalcija z kostiju te pojačana tubularna reapsorpcija kalcija.
Učinci PTH-a i vitamina D na kost su kompleksniji na što ukazuje njihov učinak kada se primjenjuju kao lijekovi. Primjena PTH-a u terapijskim dozama paradoksalno dovodi do aktivacije osteoblasta i pojačane koštane izgradnje, a terapijska primjena vitamina D također potiče izgradnju kosti.
Učinak ostalih hormona. Estrogeni kod žena u reproduktivnoj dobi imaju bitnu ulogu u procesu koštane pregradnje jer oni inhibiraju citokine koji aktiviraju osteoklaste te oponiraju učinku PTH-a. Zbog toga kod žena u menopauzi dolazi do pojave osteoporoze. Terapijska primjena gluokortikoida koči diferencijaciju osteoblasta te potiče aktivaciju osteoklasta zbog čega je osteoporoza jedna od karakterističnih nuspojava ove terapije. Kalcitonin, hormon štitne žlijezde, inhibira osteoklaste i koči koštanu razgradnju, dok u proksimalnom tubulu koči reapsorpciju kalcija i fosfata.
Laboratorijski pokazatelji koštane pregradnje. Tijekom razgradnje i izgradnje koštanoga tkiva oslobađaju se različiti enzimi i proteini koji mogu poslužiti kao biokemijski pokazatelji koštane pregradnje. U Tablici 8.26. navedeni su najznačajniji markeri koštane pregradnje.
|
Tablica 8.26. Pokazatelji koštane pregradnje
|
|
Pokazatelji koštane izgradnje
|
Pokazatelji koštane razgradnje
|
|
Alkalna fosfataza, ukupna (serum)
Alkalna fosfataza, koštano-specifična (serum)
Osteokalcin (serum)
Prokolagen tip 1 (serum)
|
Hidroksiprolin (urin)
Ukupni piridinolin (urin)
Slobodni deoksipiridinolin (urin)
Kolagen tip 1 N-telopeptid (urin)
Kolagen tip 1 C-telopeptid (serum, urin)
Koštani sijaloprotein (serum)
Tartarat-rezistentna kisela fosfataza 5b (serum)
|
Markeri koštane izgradnje. Najznačajniji markeri koštane izgradnje u kliničkoj praksi su alkalna fosfataza, serumski osteokalcin te serumski prokolagen tip 1.
Alkalna fosfataza najstariji je biokemijski pokazatelj koštane pregradnje. Postoji nekoliko izoenzima koji potiču od različitih tkiva (jetra, kosti, crijevo, slezena, bubreg, posteljica). Standardno se u serumu određuje vrijednost ukupne alkalne fosfataze, a 50 % te vrijednosti čini alkalna fosfataza porijeklom iz jetre, a 50 % porijeklom iz koštanog tkiva. U slučaju povišenih vrijednosti u razlikovanju jetrene od koštane patologije pomaže mjerenje drugih kolestatskih enzima (kod bolesti jetre osim alkalne fosfataze nalazimo i povišene vrijednosti gama-glutamil transferaze, GGT). Osim toga, danas su dostupne laboratorijske metode kojima se može određivati vrijednosti koštanoga izoenzima alkalne fosfataze.
Osteokalcin je mali protein sastavljen od 49 aminokiselina, a sintetiziraju ga u najvećoj mjeri zreli osteoblasti zbog čega se osteoklacin smatra specifičnim markerom funkcije osteoblasta. Osteokalcin pokazuje cirkadijalni ritam, tako da su ujutro njegove vrijednosti snižene i dalje se snižavaju prema sredini dana, da bi se od podneva počele povećavati i postaju najviše u vrijeme oko ponoći kada ponovno kreću padati prema jutru. Kod žena vrijednosti ovise i o menstruacijskom ciklusu tako da tijekom leutinske faze bilježimo značajno više vrijednosti. Osteoklacin je povišen u bolestima koje karakterizira visoka stopa koštane izgradnje.
Prokolagen tip 1 sadrži N-terminalni i C-terminalni produžetak koji se odstranjuju djelovanjem specifične proteaze tijekom pretvorbe prokolagena u kolagen. Laboratorijskim metodama moguće je određivati vrijednosti i C-terminalnoga peptida (P1CP) i N-terminalnoga peptida prokolagena tip 1 (P1NP). Iako oba markera imaju dobru osjetljivost u dijagnostici osteoporoze, oni se još ne upotrebljavaju rutinski u kliničkoj praksi.
Markeri koštane razgradnje. Većina biokemijskih markera koštane razgradnje su produkti enzimatske razgradnje (hidroliza) kolagena tip 1, s obzirom na to da on čini 90 % ukupnih proteina u koštanom tkivu. Danas se razvijaju i drugi, nekolagenski markeri koštane razgradnje.
Kolagenski markeri koštane pregradnje nastaju razgradnjom kolagena tip 1, a najznačajniji su ukupni hidroksiprolin, čije se vrijednosti mjere u urinu, kao i piridinolin (PYD) i deoksipiridinolin (DPD), čije su povišene vrijednosti u urinu specifičnije za koštanu patologiju. Osim toga, danas su nam na raspolaganju metode kojima se detektiraju izolirani peptidni kolageni kao što su C-telopeptid kolagena tip 1 (CTX) te N-telopeptid kolagena tip 1 (NTX).
Nekolagenski markeri koštane pregradnje su koštani sijaloprotein (BSP) koji je uključen u mineralizaciju novonastaloga koštanog matriksa i kalcifikaciju mekih tkiva te tartarat-rezistentna kisela fosfataza 5b koja se još ne koristi u kliničkoj praksi.
Poremećaji koštanoga metabolizma. U najčešće bolesti iz ove skupine ubrajamo osteoporozu, osteomalaciju i Pagetovu bolest kostiju. Uz poremećaje koštanoga metabolizma vežu se i poremećaji paratireoidne žlijezde, kao i poremećaji metabolizma kalcija što je opisano u odgovarajućim dijelovima udžbenika.
OSTEOPOROZA
Definicija. Osteoporoza predstavlja kroničnu i progresivnu bolest koštanoga sustava koja se očituje smanjenjem koštane mase i poremećajem mikroarhitekture koštanoga tkiva, a što rezultira povećanom učestalošću patoloških prijeloma. Definicija osteoporoze prema Svjetskoj zdravstvenoj organizaciji temelji sa na nalazu denzitometrije, a ona uključuje pojmove normalne koštane gustoće, osteopenije, osteoporoze i teške osteoporoze (Tablica 8.27.). Ova definicija Svjetske zdravstvene organizacije odnosi se na žene u postmenopauzi i muškarce iznad 50 godina, a ne bi se smjela primjenjivati za djecu, žene u premenopauzi i muškarce mlađe od 50 godina.
|
Tablica 8.27. Definicija osteoporoze Svjetske zdravstvene organizacije na temelju denzitometrije
|
|
Definicija
|
BMD mjerenje
|
T score
|
|
Normalna koštana gustoća
|
BMD unutar 1 SD od srednje gustoće kostiju za mlade, zdrave osobe istoga spola
|
T score ≥ -1
|
|
Osteopenija
|
BMD između 1 i 2,5 SD ispod srednje gustoće kostiju za mlade, zdrave osobe istoga spola
|
T score između -1 i -2,5
|
|
Osteoporoza
|
BMD ≥ 2,5 SD ispod srednje gustoće kostiju za mlade, zdrave osobe istoga spola
|
T score ≤ -2,5
|
|
Teška osteoporoza
|
BMD ≥ 2,5 SD ispod srednje gustoće kostiju za mlade, zdrave osobe istoga spola s anamnezom ≥1 frakture
|
T score ≤ -2,5
|
Epidemiologija. Osteoporoza predstavlja najčešći poremećaj koštanoga metabolizma i jednu od najčešćih bolesti starije životne dobi uopće te čini velik javnozdravstveni problem. Smatra se da će 50 % žena i 20 % muškaraca u dobi iznad 50 godina doživjeti bar jedan prijelom uvjetovan osteoporozom. Rizik od osteoporoze raste sa životnom dobi, a žene obolijevaju daleko češće od muškaraca (odnos 4:1). Iako se osteoporoza može javiti kod osoba svih rasa i etničkih skupina, nešto je češća pojavnost kod bijelaca, osobito sjevernoeuropskoga porijekla te Azijata. Smatra se da u Republici Hrvatskoj osteoporozu ima 35 - 40 % osoba (većinom žena) starijih od 60 godina, a osteopeniju oko 45 %. Učestalost osteoporoze je u stalnom porastu čemu pridonose produžen životni vijek, široka upotreba lijekova koji utječu na koštanu masu, suvremen način života koji uključuje ishranu siromašnu kalcijem i smanjenu fizičku aktivnost, pušenje cigareta, konzumiranje alkohola i dr.
Etiologija. Osteoporoza nastaje kao rezultat poremećene koštane pregradnje pri čemu koštana razgradnja nadmašuje koštanu izgradnju što vodi smanjenju koštane mase i poremećaju koštane mikroarhitekture. Budući da je koštana pregradnja složen proces u kojemu sudjeluju brojne molekule i mehanizmi, razumno je očekivati da brojni čimbenici mogu pogodovati nastanku osteoporoze. Na temelju etiologije načelno razlikujemo dva oblika osteoporoze: primarna i sekundarna.
Primarna osteoporoza čini više od 95 % slučajeva osteoporoze, a podrazumijeva tri podtipa: idiopatska (juvenilna) osteoporoza, postmenopauzalna osteoporoza i senilna osteoporoza (Tablica 8.28.).
|
Tablica 8.28. Osnovne karakteristike primarne osteoporoze
|
|
Tip primarne osteoporoze
|
Karakteristike
|
|
Idiopatska osteoporoza
(juvenilna osteoporoza)
|
Razvija se kod djece ili mlađih odraslih osoba
Najčešće se javlja između 8. i 14. godine
Funkcija gonada je uredna
Glavno kliničko obilježje je bol u kostima i/ili pojava patoloških fraktura
|
|
Postmenopauzalna osteoporoza
(osteoporoza tip I)
|
Javlja se kod žena nakon menopauze
Nastaje uslijed manjka estrogena
Karakterizira je ubrzan gubitak koštane mase
Najčešće posljedice su patološke frakture
|
|
Senilna osteoporoza
(osteoporoza tip II)
|
Javlja se kod žena i muškaraca starije životne dobi
Produljenjem životnoga vijeka povećava se učestalost ove bolesti
Nastaje uslijed gubitka koštane mase uvjetovanoga starenjem
Najčešće posljedice su patološke frakture
|
Sekundarna osteoporoza javlja se u manje od 5 % slučajeva, a podrazumijeva razvoj osteoporoze kao posljedice različitih bolesti i stanja koja između različitih patogenetskih mehanizama i posljedica pogađaju i metabolizam koštanoga tkiva i dovode do poremećaja koštane pregradnje. Najčešći uzroci sekundarne osteoporoze navedeni su u Tablici 8.29.
|
Tablica 8.29. Najčešći uzroci sekundarne osteoporoze
|
|
Endokrini poremećaji
|
Bolesti probavnoga sustava
|
Poremećaji koštane srži
|
|
Hipogonodizam
Hiperparatireoidizam
Hiperkorticizam
Hipertireoidizam
Preuranjena menopauza
Šećerna bolest
|
Idiopatska crijevna upalna bolest
Stanja nakon kirurških zahvata
Bolesti jetre (kolestaza)
Glutenska enteropatija
Drugi malapsorpcijski sindromi
Parenteralna prehrana
|
Multipli mijelom
Limfomi
Leukemije
Hemolitičke anmeije
Sistemska mastocitoza
Diseminirane metastaze
|
|
Bolesti veziva i nasljedne bolesti
|
Lijekovi
|
Ostale bolesti i stanja
|
|
Osteogenesis imperfekta
Ehlers-Danlosov sindrom
Marfanov sindrom
Cistična fibroza
Bolesti odlaganja glikogena
Idiopatska hiperkalciurija
|
Glukokortikoidi
Heparin
Inhibitori protnske pumpe
Ciklosporin, takrolimus
Antikonvulzivi
Litij, SSRIs
|
Kronični alkoholizam
Imobiluzacija
Reumatoidni artritis
Ankilozirajući spondilitis
Sistemski eritematozni lupus
Renalna tubularna acidoza
|
Patogeneza. Osnovni patogenetski mehanizmi koji dovode do razvoja osteoporoze su manjak estrogena, fiziološki učinak starenja na koštanu pregradnju te deficijencija kalcija i vitamina D. Osim tih glavnih mehanizama, postoje i brojni drugi, manje izraženi mehanizmi koji mogu ometati proces koštane pregradnje, a proizlaze iz osnovne bolesti ili stanja (hiperkorticizam, genetska osnova).
Manjak estrogena danas je jedan od najčešćih patogenetskih mehanizama koji dovodi do osteoporoze jer je postemenopauzalna osteoporoza općenito najčešći oblik osteoporoze. Osim fiziološkoga manjka estrogena, i različiti drugi endokrinološki poremećaji mogu dovesti do njihova deficita. Ne samo kod žena, već i kod muškaraca, manjak estrogena povezan je s gubitkom koštane mase. U odsutnosti estrogena T-stanice potiču regeneraciju osteoklasta i njihovo dulje preživljenje putem IL-1, IL-6 i TNF-α. Osim toga, u odsutnosti estrogena T-stanice inhibiraju diferencijaciju i aktivnost osteoblasta te uzrokuju njihovu preranu apoptozu, a pri tome glavnu ulogu ima IL-7. Navedene promjene dovode do prekomjerne koštane razgradnje koju ne prati odgovarajuća koštana izgradnja te u konačnici dolazi do smanjenja koštane mase.
Starenje je fiziološki proces koji ima različite reperkusije na koštani metabolizam i koštanu pregradnju. Za razliku od gubitka koštane mase pri manjku estrogena, kada ono nastaje prvenstveno zbog prekomjerne aktivacije osteoklasta i koštane razgradnje, u ovom slučaju gubitak koštane mase posljedica je slabljenja funkcije i stvaranja osteoblasta i izgradnje kosti uz normalnu razgradnju kosti. Osnovni mehanizam je gubitak i oslabljena diferencijacija mezenhimalnih matičnih stanica u osteoblaste što je fiziološka pojava u starosti, a cijelom procesu pridonosi i oslabljena apsorpcija kalcija i vitamina D. Osim toga, u starosti nalazimo i povišene vrijednosti IL-6 i TNF-α. Gubitak koštane mase povezan sa starenjem izraženiji je kod žena nego kod muškaraca.
Manjak kalcija i vitamina D u krvi dovodi do pojave sekundarnoga hiperparatireoidizma pri čemu se pojačano luči parathormon (PTH) koji manjak kalcija u krvi nastoji kompenzirati aktivacijom osteoklasta i mobilizacijom kalcija iz koštanoga tkiva.
Hiperkorticizam također je jedan od češćih uzroka osteoporoze, a može biti posljedica endokrinoloških poremećaja ili se javlja jatrogeno kao nuspojava dugotrajne kortikosteroidne terapije. Kortikosteroidi inhibiraju osteoblaste i izazivaju njihovu apoptozu što dovodi do smanjenja koštane izgradnje i gubitka koštane mase.
Genetska osnova. Iako osteoporoza nema direktan uzrok u genetskom poremećaju, polimorfizmi gena za citokine koji imaju implikacije u koštanom metabolizmu mogu biti povezani s pojavom osteoporoze. Radi se o polimorfizmima gena za Il-1, IL-6, TNF-α, kao i polimorfizmima za vitamin D receptor, koštani morfogenični protein, kolagen tip 1 i dr.
Rizični čimbenici. Na temelju navedenoga jasno se ističu glavni rizični čimbenici koji pogoduju razvoju osteoporoze: životna dob preko 50 godina, ženski spol, bijela rasa, obiteljska anamneza na osteoporozu, niži rast i smanjena tjelesna težina, amenoreja, kasna menarha, rana menopauza, postmenopauza, fizička neaktivnost i/ili imobilizacija, primjena određenih lijekova (glukokortikoidi, antikonvulzivi, heparin...), kronični alkoholizam, pušenje, hipogonodizam, deficijencija kalcija i vitamina D.
Komplikacije. Osnovna komplikacija i posljedica osteporoze je pojava patoloških fraktura. Gubitak koštane mase i poremećena mikroarhitektura kosti uzrokuje slabiju otpornost kosti na djelovanje mehaničke sile što može rezultirati prijelomom kosti i kod djelovanja slabih sila, a u težim oblicima osteoporoze prijelomi mogu nastati i bez djelovanja sile (traume), tzv. spontane frakture. Najčešće se radi o kompresivnim frakturama kralježaka, prijelomu kuka te prijeloma radijusa na tipičnoj lokaciji (loco tipico). Svjetska zdravstvena organizacija razvila je FRAX score koji pomaže u predviđanju desetogodišnjega rizika od nastanka prijeloma kosti.
Klinička slika. Osteoporoza je najčešće asimptomatska bolest. Većina oboljelih nema nikakvih simptoma sve do pojave patološkoga prijeloma. Manji broj bolesnika može imati izražen tup bol niskoga intenziteta u području donjega dijela kralježnice uslijed gubitka koštane mase. Najčešći oblik prijeloma je kompresivna fraktura kralježaka koja također može proći asimptomatski i slučajno se otkriti prilikom radiografskoga snimanja kralježnice zbog neke druge indikacije, a može se prezentirati i akutnim, jakim bolom u kralježnici ili pak pojavom dugotrajnoga tupog bola. Ovi prijelomi mogu nastati i bez jasne traume prilikom okretanja u krevetu, jakoga kašlja i sl. Moguć je i razvoj skolioze i kifoze. Prijelom kuka i podlaktice obično je povezan s traumom (pad), a praćeni su klasičnom simptomatologijom (bol, smanjena pokretljivost).
Dijagnoza. Dijagnostički se postupak sastoji od temeljite anamneze i fizikalnoga pregleda (uviđanje rizičnih čimbenika za razvoj osteoporoze), laboratorijske dijagnostike kojom obuhvaćamo rutinske pretrage te određivanje parametara koštane pregradnje, klasičnoga koštanog rendgenograma te određivanjem koštane gustoće (denzitometrija). Indikacije za mjerenje gustoće kostiju navedene su u Tablici 8.30.
Rutinske laboratorijske pretrage izvode se kod svih bolesnika sa sumnjom na osteoporozu. One imaju značaj u diferencijaciji primarne i sekundarne osteoporoze, kao i u otkrivanju uzroka sekundarne osteoporoze. Razlikovanje primarne od sekundarne osteoporoze najbolje se ispituje određivanjem ukupnoga i ioniziranog kalcija, fosfata, alkalne fosfataze, aktivnoga oblika vitamina D (25-hidroksivitamin D) te parathormona (PTH). Svi navedeni laboratorijski parametri uredni su u primarnoj osteoporozi s time da se vrijednosti alkalne fosfataze mogu povisiti nakon patoloških prijeloma nakon čega ostaju lagano povišene i kroz nekoliko mjeseci. Za otkrivanje uzroka sekundarne osteoporoze na raspolaganju nam stoje različite laboratorijske pretrage: pokazatelji jetrene i bubrežne funkcije (jetrene transaminaze, dušični metaboliti), vrijednost kalcija u 24-satnom urinu (hiperkalciurija), parathormon (hiperparatireoidizam), tireotorpni hormon (hipertireoza), testosteron i gonadotorpini (hipogonodizam), vrijednost slobodnoga kortizola u urinu i/ili prekonoćni test supresije deksametazonom (hiperkorticizam), elektroforeza proteina urina i seruma (multipli mijelom), antiglijadinska i antiendomizijska antitijela (celijakija) te biopsija koštane srži (hematološki poremećaj).
Parametri koštane pregradnje koriste se kao dopunske laboratorijske pretrage koje mogu ukazivati na poremećenu koštanu pregradnju. Osim u dijagnostici, ovi markeri imaju svoje mjesto i u praćenju terapijskoga odgovora nakon početka specifične terapije. U kliničkoj praksi najčešće se prate koštano specifična alkalna fosfataza, osteokalcin, N-telopeptid kolagena tip 1 u urinu te C-telopeptid kolagena tip 1 u serumu.
Denzitometrija je danas zlatni standard za postavljanje dijagnoze osteoporoze, a omogućava mjerenje gustoće minerala u kostima (BMD, engl. bone mineral density). Postoji više oblika (metoda) denzitometrije, a najčešće se koristi DXA metoda (engl. dual x-ray absorptiometry) pri čemu se kroz kost propuštaju dvije X-zrake u malim dozama, a iza kosti nalazimo senzor koji mjeri dozu X-zraka koje su prošle kroz kost. Na temelju razlike energije propuštenih i apsorbiranih X-zraka procjenjuje se gustoća minerala kosti (BMD) te se izražava u jedinici g/cm2. Snimanje se obično vrši u području lumbalne kralježnice, kuka ili distalnoga radijusa, a zračenje koje se primjenjuje tijekom pretrage je minimalno (ekvivalent zračenju koje se primi tijekom leta avionom u trajanju od 3 do 9 sati). Rezultati pretrage izražavaju se na tri načina: 1) apsolutnim brojem (g/cm2); 2) kao "T score" koji predstavlja odstupanje izmjerene vrijednosti BMD-a od vršne koštane mase mladih osoba izraženo u standardnim devijacijama (SD) te 3) kao "Z score" koji predstavlja odstupanje izmjerene vrijednosti BMD-a od prosječne koštane mase osoba iste dobi izraženo u standardnim devijacijama (SD). T-score koristi se za izražavanje BMD-a postmenopauzalnih žena i muškaraca starijih od 50 godina, dok se Z-score koristi za izražavanje BMD-a žena u reproduktivnoj dobi i muškaraca mlađih od 50 godina. Pri tumačenju nalaza treba imati na umu da koštane promjene kao što su deformiteti, osteofiti i slične promjene mogu lažno povisiti BMD, osobito pri snimanju lumbalne kralježnice zbog čega se kod starijih bolesnika (> 65 godina) preporuča skeniranje kuka. Tumačenje vrijednosti T-scorea prikazano je u Tablici 8.27. u definiciji bolesti i u toj kategoriji bolesnika ona je dovoljna za postavljanje dijagnoze osteoporoze. Kod interpretacije Z-scorea u dijagnostici osteoporoze, vrijednosti koje su niže od -2,0 SD definiraju se kao vrijednosti ispod očekivanoga raspona za dob, a vrijednosti koje su iznad -2,0 SD kao vrijednosti unutar očekivanoga raspona za dob. Za tu kategoriju bolesnika osteoporoza se ne može dijagnosticirati samo na temelju nalaza denzitometrije, nego u obzir treba uzeti i druge dijagnostičke elemente. Neki uređaji za izvođenje DXA-e omogućavaju prikaz kralježnice iz latero-lateralne projekcije i to od područja četvrtoga torakalnog do četvrtoga lumbalnog kralješka pri čemu se može izvršiti i procjena prijeloma kralježaka što se inače standardnim antero-posteriornim snimanjem ne može. Ovaj dodatak naziva se VFA (engl. vertebral fracture assessment).
Ostale metode mjerenja gustoće kostiju rjeđe se primjenjuju u kliničkoj praksi, a uključuju kvantitativnu kompjuteriziranu tomografiju (QCT), kvantitativni ultrazvuk te magnetnu rezonancu. Iako kvantitativni CT (QCT) može bolje i objektivnije procijeniti BMD od DXA-e, rijetko se koristi u kliničkoj praksi zbog daleko veće doze zračenja, cijene same pretrage te manjkavosti podataka koje potvrđuju njegovu sposobnost predviđanja rizika od prijeloma. Kvantitativni ultrazvuk koristi se u mjerenju koštane mase na temelju izračunavanja prigušenja ultrazvučnoga vala tijekom prolaska kroz kost. Pretraga se najčešće izvodi na petnoj kosti, a svi patološki rezultati moraju se provjeriti drugom metodom stoga se danas vrlo rijetko upotrebljava.
|
Tablica 8.30. Indikacije za mjerenje gustoće kostiju (BMD)
|
- Žene u dobi iznad 65 godina i muškarci iznad 70 godina, bez obzira na prisutnost rizičnih čimbenika
- Žene u ranoj postmenopauzi te muškarci u dobi između 50 i 69 godina ako su prisutni neki rizični čimbenici
- Žene i muškarci u ranijoj životnoj dobi sa sumnjom na patološki prijelom uz prisutnost rizičnih čimbenika
|
Klasični koštani rendgenogram koristan je u dijagnostici osteoporoze, ali ne kao osnovna, nego dopunska metoda. Indiciran je kod bolesnika sa sumnjom na patološki prijelom, a osim prijeloma može ukazivati na osteopeniju i smanjenu koštanu gustoću koja upućuju na osteoporozu.
FRAX score (engl. fracture risk assessment tool) predstavlja individualni izračun desetogodišnjega rizika od nastanka osteoporotične frakture kuka ili druge osteoporotične frakture. Rizik se računa na temelju unosa podataka o dobi, spolu, T-scoru te drugih rizičnih čimbenika, a rezultat se dobiva matematičkim izračunom u obliku dva postotka (postotak koji nam govori o riziku nastanka frakture kuka te postotak koji nam govori o riziku nastanka drugih osteoporotičkih fraktura). FRAX score razvila je Svjetska zdravstvena organizacija.
Diferencijalna dijagnoza. Diferencijalno dijagnostički u obzir dolaze sljedeće bolesti: prolaps intervertebralnoga diska, prethodna trauma, maligniteti (metastaze), multipli mijelom, osteomalacija, Pagetova bolest, hiperparatiroidizam (primarni i sekundarni), osteogenesis imperfecta.
Liječenje. Terapijske mjere u liječenju osteoporoze obuhvaćaju nefarmakološko, farmakološko te kirurško liječenje. Cilj liječenja je spriječiti gubitak koštane mase da bi se spriječio nastanak prvoga prijeloma te izbjegavanje novih prijeloma kod osoba koje su već imale prijelom.
Nefarmakološko liječenje osteoporoze uključuje provođenje općih mjera liječenja kao što su uklanjanje rizičnih čimbenika, održavanje tjelesne aktivnosti i provođenje vježbi te prikladna prehrana.
Uklanjanje rizičnih čimbenika podrazumijeva redukciju prisutnih čimbenika rizika koji se mogu modificirati te uklanjanje čimbenika koji povećavaju rizik od pada kod starijih osoba (sprečavanje pojave fraktura). Redukcija prisutnih čimbenika rizika podrazumijeva prestanak pušenja, smanjenje unosa kofeina, umjereno konzumiranje alkohola, odgovarajuću ishranu, izbjegavanje dugotrajnih dijeta bez unošenja proteina, povećanje tjelesne težine ako je BMI < 20 kg/m², održavanje fizičke aktivnosti te racionalnu upotrebu lijekova koji utječu na razvoj osteoporoze. Redukcija čimbenika rizika koji povećavaju rizik od pada kod starijih osoba uključuje korekciju slaboga vida, izbjegavanje uporabe hipnotika s dugim djelovanjem, izbjegavanje rada na visini, korištenje obuće s niskim i širokim potpeticama te korištenje pomagala pri kretanju (štap i dr.).
Vježbe i tjelesna aktivnost. Fizička aktivnost značajna je u ranom djetinjstvu da bi se dosegla genetski određena maksimalna gustoća koštane mase koja će odgoditi pojavu osteoporoze (rana prevencija). Idealno je nastaviti s vježbama (samo ciljane vježbe s opterećenjem imaju učinak na skelet) tokom cijeloga života za održavanje koštane mase i jačanja muskulature. Bilo koja fizička aktivnost smanjuje rizik od kardiovaskularnih bolesti (plivanje, biciklizam i brzi hod), a samo jače vježbe s opterećenjem i otporom dovode do smanjenja rizika od osteoporoze. Kod već prisutne osteoporoze potrebno je prilagoditi vježbe svakom bolesniku prema životnom dobu i stanju kralježaka (frakture).
Prehrana. Pravilna ishrana značajna je u svakom stadiju prevencije i liječenja osteoporoze. To se prije svega odnosi na unos kalcija i vitamina D. Zadovoljavajući unos vitamina D je od 600 do 800 ij dnevno, a izlaganje sunčevoj svjetlosti od 15 minuta dnevno dovoljno je da bi se omogućila endogena sinteza vitamina D.
Farmakološko liječenje osteoporoze indicirano je kod sljedećih bolesnika: 1) bolesnici s potvrđenom osteoporozom na temelju nalaza denzitometrije (DXA T-score ispod -2,5 SD); 2) bolesnici s osteopenijom (DXA T-score između -2,5 i -1,0) koji imaju FRAX score veći od 3 % za prijelom kuka i/li veći od 20 % za drugi osteoporotični prijelom te 3) žene s prethodnom frakturom kuka ili kralježaka udruženom s minimalnom traumom. Pri farmakološkom liječenju na raspolaganju je nekoliko skupina lijekova: vitamin D i kalcij, spolni hormoni, bisfosfonati, denosumab (monoklonalno protutijelo), teriparatid (sintetski parathormon), selektivni modulatori estrogenskih receptora te kalcitonin. Oni mogu djelovati tako da smanjuju koštanu razgradnju (antiresorptivni lijekovi) ili da potiču koštanu izgradnju (osteoanabolici).
Kalcij i vitamin D. Iako je manjak vitamina D i kalcija odgovoran prvenstveno za nastanak osteomalacije, i kod bolesnika sa osteoporozom potrebno je osigurati dostatan unos tih nutrijenata jer su prijeko potrebni za odvijanje normalne koštane pregradnje. Dnevne potrebe za vitaminom D u dozi od 600 do 800 ij teško je osigurati putem hrane ili sunčanja (osobito tijekom zimskih mjeseci) pa se preporuča njihova suplementacija u obliku kolekalciferola koji se daje peroralno u dozi od 800 do 2000 ij/dan (preporuka je održavati serumske vrijednosti 25-hidroksivitamina D iznad 30 - 50 ng/mL). Također se preporuča i unos kalcija od najmanje 1000 mg/dan kod svih osoba, a za postmenopauzalne žene i muškarce iznad 70 godina i 1200 mg/dan. Većina osoba osigurava ove potrebe unosom kalcija putem hrane te se suplementacija preporuča samo kod bolesnika s malapsorpcijskim sindromom ili u slučaju nedovoljnoga unosa putem hrane. Kalcij se može primjenjivati u obliku kalcij citrata ili kalcij karbonata. Pojačan rizik od razvoja kardiovaskularnoga incidenta kod bolesnika koji dodatno uzimaju kalcij nije potvrđen, ali je zabilježena pojačana pojavnost nefrolitijaze.
Spolni hormoni. Primjena spolnih hormona kod bolesnika s hipogonadizomom, bilo da se radi o muškarcima, bilo o ženama, dokazano smanjuje učestalost osteoporoze. Kod žena se primjenjuje kombinacija estrogena i progresterona, iako sam učinak na kost ostvaruju estrogeni, dok se progesteron dodaje u svrhu prevencije nastanka hiperplazije i karcinoma endometrija. Prevencija osteoporoze kod muškaraca s hipogonadizmom ostvaruje se primjenom testosterona. Primjena ovih hormona opisana je u odgovarajućim dijelovima udžbenika.
Bisfosfonati su među najčešće primjenjivanim antiresorptivnim lijekovima. Svoj antiresorptivni učinak ostvaruju djelovanjem na osteoklaste (inhibicija vezanja osteoklasta za koštani matriks, apoptoza). Primjena ovih lijekova dokazano povećava koštanu gustoću i smanjuje rizik od nastanka patoloških prijeloma. Danas osim peroralnih oblika bisfosfonata postoje i oni koji su namijenjeni za intravensku primjenu. Kako bi se poboljšala peroralna apsorpcija lijeka, preporuča se uzimanje lijeka s dosta vode najmanje 40 minuta prije obroka, a kako bi se spriječio nastanak ezofagitisa preporuča se izbjegavati ležeći položaj nekoliko sati nakon uzimanja lijeka. Doze i posebne napomene uz pojedine bisfosfonata navedene su u Tablici 8.31. Bisfosfonati se izlučuju putem bubrega, ali se njihova doza ne mora smanjivati sve dok je klirens kreatinina iznad 35 mL/min. Najčešće nuspojave peroralnih bisfosfonata su mučnina, povraćanje, bolovi u prsima te promuklost, a povećan je i rizik od razvoja erozivnoga ezofagitisa, osobito kod bolesnika s hijatalnom hernijom i gastroezofagealnim refluksom. Intravenska primjena bisfosfonata može uzrokovati tzv. odgovor akutne faze koji uključuje vrućicu, zimicu i tresavicu, mišićno-koštani bol, mučninu, povraćanje, proljev, dispneju te glavobolju. Simptomi mogu trajati do nekoliko dana i obično spontano prestaju, a dobro reagiraju i na primjenu nesteroidnih protuupalnih lijekova ili paracetamola te antihistaminika (loratadin). Osteonekroza donje ili gornje čeljusti rijetka je komplikacija primjene bisfosfonata, a obično se javlja nakon ekstrakcije zuba. U više od 95 % slučajeva povezana je s intravenskom primjenom bisfosfonata, osobito kod bolesnika s multiplim mijelomom ili koštanim metastazama, dok je vrlo rijetko opisana kod bolesnika koji su na peroralnoj terapiji. Rizik od prijeloma potrebno je ponovno procijeniti nakon 3 do 5 godina, te kod žena koje i dalje imaju visok rizik od prijeloma nastaviti terapiju, dok kod onih s niskim do umjereno niskim rizikom treba razmotriti tzv. „drug holiday“, odnosno privremenu stanku od uzimanja lijekova.
|
Tablica 8.31. Peroralni i parenteralni bisfosfonati
|
|
Peroralni bisfosfonati
|
Intravenski bisfosfonati
|
|
Alendronat
|
Risedronat
|
Ibandronat
|
Zoledronat
|
Pamidronat
|
|
70 mg tbl
1 x tjedno
|
75 mg tbl
1 x tjedno
|
150 mg tbl
1 x mjesečno
|
2 - 3 mg iv
1 x godišnje
|
30 - 60 mg iv
2 - 4 x godišnje
|
|
Alendronat i risedronat smanjuju rizik od nastanka vertebralnih i nevertebralnih prijeloma s naglaskom da je alendronat superironiji u prevenciji nevertebralnih prijeloma. Ibandronat smanjuje rizik samo od vertebralnih prijeloma.
|
Primjenjuju se samo kod bolesnika koji ne mogu uzimati peroralne bisfosfonate. Daju se u obliku sporih intravenskih infuzija (kroz 15 do 30 minuta).
|
Denosumab je monoklonalno protutijelo koje inhibira proliferaciju i sazrijevanje osteoklasta te na taj način ostvaruje svoj antiresorptivni učinak. Lijek se primjenjuje supkutano u dozi od 60 mg svakih 6 mjeseci. Kod oko 10 % bolesnika nakon primjene se razvijaju simptomi nalik gripi, ali ukupno gledano, dobro se podnosi. Od ostalih nuspojava spominju se najčešće hipokalcijemija, a rijetko hiperkolesterolemija, ekcem i dermatitis, pojačana sklonost infekcijama te pankreatitis. Dozu nije potrebno prilagođavati bubrežnoj funkciji i može se davati i bolesnicima s uznapredovalom bubrežnom bolesti. Denosumab se obično primjenjuje kroz 3 do 5 godina. Učinci denosumaba na pregradnju kostiju su reverzibilni, te ako se lijek ne aplicira na vrijeme dolazi do pojačane koštane razgradnje što može rezultirati tzv. „rebound fenomenom“ odnosno povećanom incidencijom vertebralnih fraktura. Dakle, drug holiday ili nagao prestanak liječenja se ne preporučuje, tj. ako se lijek prekida, potrebno je nastaviti liječenje drugim lijekom. Rizik od prijeloma procjenjuje se nakon 5 do 10 godina, ako je visok - nastavlja se liječenje denosumabom ili drugom vrstom terapije. Denosumab smanjuje rizik od nevetebralnih i vertebralnih fraktura, kao i fraktura kuka.
Teriparatid i abaloparotid su analozi paratireoidnoga hormona koji djeluju kao osteoanabolici, tj. stimuliraju stvaranje novoga koštanog tkiva ako se primjenjuju intermitentno, za razliku od fiziološkoga učinka PTH-a koji uzrokuje koštanu razgradnju. Zbog pojačane koštane izgradnje bolesnici koji uzimaju ovaj lijek moraju imati odgovarajuću suplementaciju kalcija i vitamina D. Lijek se primjenjuje supkutano u dozi od 20 mg/dan kroz dvije godine i dokazano poboljšava koštanu masu u većini kostiju s iznimkom distalnoga radijusa. Zbog povećanoga rizika od nastanka osteosarkoma koji je dokazan samo na animalnim modelima, on se mora izbjegavati kod bolesnika koji nose rizik za razvoj tog malignoma: Pagetova bolest, povišene vrijednosti alkalne fosfataze nejasne etiologije, ranija radioterapija koštanoga tkiva te osobna anamneza na koštane tumore. Najčešće nuspojave ovoga lijeka su alergijske reakcije na području mjesta aplikacije, ortostatska hipotenzija, artralgije, mišićni grčevi, hiperkalcijemija, depresija te pojačana učestalost pneumonija. Nakon završene terapije teriparatodom preporuča se nastavak liječenja bisfofonatima ili denosumabom kako bi se zadržala stvorena koštana masa. U težim oblicima bolesti opravdana je i istodobna primjena teriparatida i denosumaba. Ovi lijekovi smanjuju rizik od nevertebralnih i vertebralnih fraktura, ali ne i fraktura kuka.
Selektivni modulatori estrogenskih receptora (SERMs) su lijekovi koji imaju ulogu u prevenciji nastanka osteoporoze, dok imaju vrlo malen učinak na samo liječenje osteoporoze. Svoj farmakološki učinak ostvaruju imitiranjem učinka estrogena na koštano tkivo. Najpoznatiji lijek iz ove skupine je raloksifen koji se primjenjuje u dozi od 60 mg/dan per os. Konačni učinak raloksifena slabiji je od učinka samoga estrogena. Primjena ovoga lijeka nije povezana s smanjenjem pojavnošću valunga, hiperplazijom endometrija, karcinoma uterusa, adneksa i dojke (dokazano smanjuje rizik od nastanka dojke za 70 %), no kao i kod primjene estrogena, ostaje povišen rizik od nastanka tromboembolijskih incidenata. Raloksifen također smanjuje vrijednosti LDL kolesterola, ali ne utječe na vrijednosti HDL kolesterola, kao ni na aterosklerotske plakove. SERMs imaju učinak samo na vertebralne frakture, a preporučuju se ženama koje su mlađe od 60 godina, odnosno koje su u menopauzi manje od 10 godina.
Kalcitonin danas ima nizak značaj u liječenju osteoporoze pored ostalih, prethodno navedenih lijekova. On djeluje antiresorptivno inhibirajući osteoklaste. Danas se koristi jedno kod bolesnica koje imaju visok rizik od prijeloma te ne podnose navedene lijekove, a može se još koristiti, iako rijetko, za liječenje akutnoga bola nakon patoloških prijeloma kralježaka zbog svoga analgetskog učinka. Analgetski učinak postiže kroz nekoliko tjedana i traje do nekoliko mjeseci. Najčešće se koristi u obliku raspršivača (salmon kalcitonin) koji se aplicira nazalno preko sluznice nosa, a primjenjuje se u dozi od 200 ij/dan (jedan potisak u nosnicu). Najčešće nuspojave su iritativni iritis i epistaksa. Kalcitonin dokazano smanjuje i rizik od verterbralnih prijeloma, ali u prevenciji drugih osteoporotičnih prijeloma njegov učinak nije dokazan.
Romosozumab je predstavnik nove klase lijekova, radi se o monoklonskom protutijelu koje inhibira sklerostin čija je funkcija inibicija Wnt signalnoga puta koji ima značajnu ulogu u razvoju kostiju, homeostazi koštanoga metabolizma odraslih te pregradnji kostiju. Učinak romosoumaba je jedinstven: istovremeno povećava stvaranje kostiju i smanjuje resorpciju kostiju. Povećava mineralnu gustoću kostiju i u lumbalnoj kralježnici i u ukupnom kuku te u trabekularnoj i kortikalnoj kosti, što dovodi do povećanja čvrstoće kostiju i smanjenoga rizika od vertebralnih i nevertebralnih prijeloma, kao i prijeloma kuka. Preporučen je za liječenje žena u postmenopauzi s osteoporozom s vrlo visokim rizikom od prijeloma, poput onih s više prijeloma kralježaka u trajanju do jedne godine. Aplicira se jednom mjesečno supkutano. Međutim, osobe s visokim rizikom od kardiovaskularnih bolesti i moždanoga udara ne smiju ga uzimati zbog povećane incidencije kardiovaskularnih događaja.
Kirurško liječenje rezervirano je za zbrinjavanje nastalih patoloških prijeloma s ciljem olakšanja simptoma, prevencije značajnije invalidnosti te unapređenja kvalitete života. Perkutana vertebroplastika i kifoplastika danas su najčešći zahvati kod bolesnika s osteoporozom.
Preporuke za liječenje osteoporoze prema smjernicama Britanske nacionalne skupine za osteoporozu (The National Osteporosis Guideline Group, NOGG) iz 2019. godine:
- Farmakološko liječenje preporuča se kod žena u postmenopauzi s velikim rizikom od prijeloma, posebno onih koje su imale nedavni prijelom jer koristi nadmašuju rizike.
- Kod žena u postmenopauzi s visokim rizikom od prijeloma preporuča se početno farmakološko liječenje bisfosfonatima (alendronat, risedronat, zoledronska kiselina, ibandronat) kako bi se smanjio rizik od prijeloma. Ibandronat se ne preporučuje za smanjenje nevertebralnih prijeloma i prijeloma kuka.
- Kod žena u postmenopauzi s osteoporozom te visokim rizikom od osteoporotskih prijeloma, preporuča se denosumab kao alternativni početni tretman.
- Kod žena u postmenopauzi s osteoporozom te vrlo visokim rizikom od prijeloma, poput onih s teškim ili višestrukim prijelomima kralježaka, preporuča se liječenje teriparatidom ili abaloparatidom do 2 godine.
- Kod žena u postmenopauzi s osteoporozom i visokim rizikom od prijeloma i sa sljedećim karakteristikama: nizak rizik od duboke venske tromboze, bisfosfonati ili denosumab nisu prikladni te visoki rizik od raka dojke - preporuča se raloksifen za smanjenje rizika od prijeloma kralježaka.
- Kod žena u postmenopauzi s visokim rizikom od prijeloma i sljedećim karakteristikama: mlađi od 60 godina ili 10 godina nakon menopauze, nizak rizik od duboke venske tromboze, bisfosfonati ili denosumab nisu prikladni, postojanje vazomotornih simptoma, bez prethodnoga infarkta miokarda ili moždanoga udara te bez raka dojke, preporuča se hormonska nadomjesna terapija menopauze uz upotrebu estrogena samo kod žena s histerektomijom, kako bi se spriječile sve vrste prijeloma.
- Kod žena u postmenopauzi s niskom koštanom gustoćom i visokim rizikom od prijeloma kalcij i vitamini D potrebno je koristi kao dodatak terapiji osteoporoze.
- Preporuča se praćenje BMD-a (engl. bone mieral density) svake 1 - 3 godine kako bi se procijenio učinak terapije.
Prognoza. Sve epidemiološke mjere danas su usmjerene na ranu dijagnostiku osteoporoze i poduzimanje preventivnih mjera. Osteoporoza ima progresivni tijek koji može usporiti primjena lijekova i bolesnikovo pridržavanje samoga liječenja. Primjenom lijekova i pridržavanjem nefarmakoloških mjera danas se u velikoj mjeri može izbjeći nastanak osteoporotičnih prijeloma.
OSTEOMALACIJA
Definicija. Osteomalacija je naziv za poremećaj koštanoga metabolizma kod odraslih osoba kojega karakterizira poremećaj mineralizacije novonastaloga organskoga koštanog matriksa pri čemu nastaje kost smanjene čvrstoće sklona deformitetima i prijelomima.
Etiopatogeneza. Osteomalacija se razvija uslijed manjka ili poremećaja metabolizma minerala koji sudjeluju u koštanoj izgradnji (kalcij, fosfat), kao i zbog poremećaja hormona koji sudjeluju u tom procesu (vitamin D, paratireoidni hormon). Osim toga, osteomalacija može nastati i zbog prisutnosti inhibitora mineralizacije, kao i u nekim bolestima koštanog matriksa (Tablica 8.32.).
|
Tablica 8.32. Etiologija osteomalacije
|
|
Manjak vitamina D
|
Manjak kalcija i fosfata
|
Ostali uzroci
|
|
Malnutricija (smanjen unos hranom)
Smanjena izloženost svjetlosti
Malapsorpcijski sindrom
Kronična bubrežna bolest
Kronična bolest jetre
Antikonvulzivi
|
Malnutricija (smanjeni unos hranom)
Malapsorpcijski sindrom
X-vezana hiposfosfatemija
Sporadična hipofosfatemija
Kronična bubrežna bolest
Primjena antacida koji vežu fosfate
|
Inhibitori mineralizacije (aluminij, bisfosfonati, fluoridi)
Hipofosfatazija
Osteogeneis imperfecta
Aksijalna osteomalacija
Onkogena osteomalacija
|
Manjak vitamina D najčešći je uzrok osteomalacije u kliničkoj praksi. Uslijed manjka aktivnoga oblika vitamina D dolazi do smanjenja apsorpcije kalcija iz probavnoga sustava zbog čega se razvija hipokalcijemija i posljedično pojačano lučenje parathormona (hiperparatireoidizam). Parathormon potiče mobilizaciju kalcija iz kosti te potiče lučenje fosfata putem bubrega zbog čega se razvija hipofosfatemija. Mobilizacija kalcija iz kosti uz razvijenu hipofosfatemiju dovodi do poremećaja mineralizacije kosti i razvoja osteomalacije.
Klinička slika. Bolest ima dug asimptomatski period, a prvi simptomi su difuzni, slabo lokalizirani bolovi u kostima, osobito u donjem dijelu leđa i natkoljenicama te slabost proksimalnih skupina mišića što bolesnike ograničava u njihovim svakodnevnim aktivnostima (hodanje uz stepenice, dizanje predmeta, ustajanje i uspravljanje i sl.). Postupno se razvijaju deformacije (kifoza, skolioza, genua vara et valga), a bolesnici su i skloniji nastanku prijeloma uslijed male traume ili bez ikakve traume.
Dijagnoza se postavlja na osnovi anamneze i fizikalnoga pregleda, laboratorijskih nalaza, radiološke obrade, a u nejasnim situacijama indicirana je i biopsija kosti. Laboratorijski nalazi uključuju određivanje vrijednosti kalcija, fosfata, vitamina D (25-hidroksivitamin D3, 1,25-dihidroksikolekalciferol) te parathormona. Nalazi ovise o etiologiji bolesti. U osteomalaciji uslijed manjka vitamina D nalazimo hipokalcijemiju, hipofosfatemiju, sniženu razinu 25-hidroksivitamina D3 te povišene vrijednosti parathormona i alkalne fosfataze. Vrijednosti 25-hidroksivitamina D3 mogu biti uredne kod razvoja sekundarnoga hiperparatireoidizma. Radiološki nalaz može biti nespecifičan i ukazivati samo na smanjenu gustoću kostiju, a rijetko se uviđaju tzv. Looserove zone, tj. bilateralne, tanke, radiolucentne (prozračne) trake koje se pružaju okomito na periost, što se obično viđa na rebrima, zdjeličnim kostima, vratu femura te vanjskom rubu lopatica. Ove se promjene smatraju patognomoničnim za osteomalaciju. Denzitometrija također ukazuje na smanjenu gustoću kostiju. U nejasnim situacijama preporuča se biopsija kosti.
Liječenje. Osnovu liječenje čini liječenje osnovne bolesti koja je u podlozi osteomalacije. Primjena vitamina D3, kalcija i fosfata čini osnovnu farmakološku mjeru u liječenju osteomalacije. Suplementacija vitamina D3, kalcija i fosfata treba biti usklađena s laboratorijskim nalazima.
PAGETOVA BOLEST KOSTIJU (OSTEITIS DEFORMANS)
Definicija. Pagetova bolest kostiju predstavlja kronično koštano oboljenje koje karakterizira lokaliziran poremećaj koštane pregradnje koji može zahvatiti jednu ili više kostiju, a sastoji se od tri razvojne faze: osteoklastična, osteoblastična te osteosklerotična faza.
Epidemiologija. Pojavnost Pagetove bolesti pokazuje specifičnu geografsku raspodjelu pri čemu se primjećuje visoka učestalost bolesti u zemljama zapadne i srednje Europe s iznimkom skandinavskih zemalja, dok je učestalost kod domorodačkoga stanovništva Amerike, Afrike, Azije i Australije niska. Pojavnost bolesti u tim područjima uglavnom je povezana s migracijom stanovništva s područja visoke učestalosti bolesti. Bolest se nešto češće javlja kod muškaraca nego kod žena, najčešće oko četrdesete godine života i dalje raste s povećanjem dobi. Pravu incidenciju bolesti teško je procijeniti jer velik dio oboljelih nema jasna klinička očitovanja i ostaje nedijagnosticiran.
Etiologija. Točan uzrok i mehanizam bolesti nisu poznati. Bolest počinje prekomjernom aktivacijom osteoklasta zbog čega dolazi do ubrzane i povećanje razgradnje kosti nakon čega slijede faza pojačane koštane izgradnje pri čemu nastaje novo koštano tkivo poremećene građe i strukture. Kao mogući pokretački mehanizmi ovih promjena spominju se genetske promjene te virusna infekcija.
Genetska podloga bolesti. O mogućem utjecaju genetike na pojavnost bolesti sugerira činjenica da se kod više od 40 % oboljelih ista bolest otkrije kod najbližih srodnika. Do sada je otkriveno više gena čije mutacije mogu imati utjecaja na pojavnost Pagetove bolesti, a svi su oni povezani s RANKL/RANK/OPG sustavom: TNFRSF11A gen, TNFRSF11B gen, SQSTM1 gen te VCP gen. Istraživanja pojavnosti bolesti unutar obitelji ukazuje većinom na autosomno-dominantni način nasljeđivanja.
Virusna infekcija. Na virusnu infekciju kao mogući pokretački mehanizam bolesti ukazuje histološki nalaz nuklearnih i citoplazmatskih inkluzija koje nalikuju na inkluzije kakve nalazimo kod infekcija uzrokovanih paramiksovirusima, iako ta teorija nije do kraja razjašnjena i dokazana.
Patogeneza. U patogenezi Pagetove bolesti kostiju razlikujemo tri faze (stadija): osteoklastična, osteoblastična i osteosklerotična faza. Prvo se javlja osteoklastična faza koju karakterizira pojačana aktivnost osteoklasta, pojačan gubitak koštanoga tkiva te hipervaskularizacija. Aktivirani osteoklasti poprimaju svoj klasičan izgled pri čemu postaju uvećani (i do 10 puta u odnosu na svoju klasičnu veličinu) te sadržavaju povećan broj jezgara. Druga faza bolesti je osteoblastična faza koju karakterizira pojačana aktivnost osteoblasta i stvaranje nove kosti, a dolazi i do propadanja koštane srži koja se zamjenjuje vezivnim tkivom. S vremenom aktivnost osteblasta nadmašuje aktivnost osteoklasta. U trećoj fazi bolesti, osteosklerotičnoj fazi, aktivnost osteoklasta značajno slabi, a u bolesti dominira povećana količina novostvorenoga koštanog tkiva poremećene strukture, raspoređenoga po nepravilnim nakupinama koje su međusobno odijeljene cementnim linijama (slika mozaicizma). Osim toga, kost sadrži bogatu vaskularnu mrežu i dosta fibroznoga tkiva. U istome trenutku u području zahvaćene kosti možemo nalaziti promjene koje odgovaraju svim trima fazama bolesti.
Klinička slika. Promjene na kostima dugo su asimptomatske i bolest se obično klinički prezentira nakon četvrtoga ili petog desetljeća života. Bolest može zahvati samo jednu kost (monoostotički oblik) ili više njih (poliostotički oblik). Najčešće zahvaćene kosti su zdjelične kosti, lumbalni kralješci, natkoljenična kost, lubanja i goljenična kost.
Koštani simptomi. Najčešći simptomi su bol i deformitet u području zahvaćene kosti. Deformiteti najviše dolaze do izražaja kada se radi o dugim, cjevastim kostima (stav nogu kao kauboj) ili lubanji (znak tijesnoga šešira). Češća je pojavnost patoloških prijeloma koji dovode do svojih klasičnih očitovanja.
Izvankoštani simptomi. Kompresivne neuralgije najčešća su izvankoštana očitovanja ove bolesti, a nastaju uslijed pritiska novostvorene kosti na živce koji se nalaze u neposrednoj blizini, a klinička slika ovisi o zahvaćenom živcu. Uslijed pojačane koštane pregradnje i oslobađanja kalcija češća je pojavnost hiperkalcijemije, patoloških kalcifikacija i nefrolitijaze. U izraženim oblicima bolesti moguć je i razvoj srčanoga popuštanja s povećanim udarnim volumenom (engl. high-output cardiac failure) uslijed velikoga broja arteriovenskih fistula u području novonastaloga koštanog tkiva.
Osteosarkom. Bolesnici s Pagetovom bolesti kostiju imaju i do trideset puta veći rizik od razvoja osteosarkoma u odnosu na osobe bez te bolesti.
Dijagnoza. Pagetova bolest kostiju dijagnosticira se na osnovi anamneze, fizikalnoga pregleda s naglaskom na pregled koštano-zglobnoga sustava, laboratorijskih pretraga te specifičnih slikovnih metoda. Najčešće se dijagnoza postavlja kod asimptomatskih bolesnika prilikom obrade slučajno nađene povišene vrijednosti alkalne fosfataze u serumu ili patološkoga radiološkog nalaza na kostima.
Laboratorijski nalazi. Kod ovih bolesnika karakteristično su povišeni markeri koštane pregradnje, a među njima se ističu vrijednosti alkalne fosfataze, iako i one mogu biti uredne ako se radi o zahvaćenosti manjega segmenta jedne kosti. Posljedično pojačanoj koštanoj razgradnji, u prvim fazama bolesti nalazimo hiperkalcijemiju i hiperkalciuriju, iako se ono viđa rjeđe ako nalazimo opsežne koštane promjene.
Klasični rendgenogram zahvaćenih kostiju može ukazivati na osteolitičke i osteoskleroične promjene te se koristi kao inicijalna pretraga u dijagnostičkoj obradi bolesnika. Sama za sebe, bez kliničkih i laboratorijskih parametara, ne može ukazivati na dijagnozu Pagetove bolesti, ali pruža uvid u opseg patoloških promjena, a bitan je i u dijagnostici patoloških prijeloma.
Scintigrafija kosti manje je specifična, ali je osjetljivija metoda za otkrivanje aktivnih koštanih lezija u odnosu na klasični rendgenogram. Glavnu diferencijalno-dijagnostičku dilemu kod nalaza aktivnih koštanih lezija kod ovih bolesnika predstavljaju koštani sekundarizmi (osobito kod karcinoma prostate).
Biopsija kosti je indicirana kod svih bolesnika kod kojih dijagnoza ostane nejasna nakon obavljenih prethodno navedenih pretraga, a osobito ako se sumnja na mogućnost neoplastičnoga procesa.
Liječenje. Asimptomatski bolesnici s ograničenim koštanim promjenama ne zahtijevaju specifično liječenje nego samo redovne kontrole i praćenje. Liječenje je indicirano za sve simptomatske bolesnike, kao i za asimptomatske s opsežnim koštanim promjenama (osobito lubanje, dugih kostiju i kralježaka). Praćenje bolesnika uključuje redovne kliničke preglede i serijsko mjerenje alkalne fosfataze koju koristimo kao marker aktivnosti bolesti te nam služi za procjenu terapijskoga učinka.
Farmakološko liječenje. Osnovu liječenja čini primjena bisfosfonata koji svojim antiresorptivnim učinkom mogu spriječiti daljnji razvoj bolesti, osobito ako se primijene u što ranijoj fazi. Lijek izbora je intravenski bisfosfonat – zoledronat koji primijenjen u jednoj dozi od 5 mg normalizira vrijednosti alkalne fosfataze u roku od 6 mjeseci kod više od 90 % bolesnika. Drugi izbor je peroralna primjena risedronata u dozi od 30 mg/dan kroz dva mjeseca ili alendronata u dozi od 40 mg/dan kroz šest mjeseci, ali je postotak bolesnika s normalizacijom alkalne fosfataze daleko manji. Prije početka terapije bisfosfonatima treba prekontolirati vrijednosti kalcija i vitamina D te osigurati dostatnu suplementaciju tijekom liječenja. Najčešće nuspojave bisfosfonata opisane su ranije u dijelu o liječenju osteoporoze.
Kirurško liječenje rezervirano je za zbrinjavanje patoloških prijeloma, korekcije deformiteta i liječenje komplikacija (dekompresija živaca i sl.). Prije samoga zahvata preporuča se liječenje bisfosfonatima kako bi se smanjila vaskularizacija kosti i preveniralo obilno krvarenje tijekom zahvata.
Prognoza. Ukupno gledano, bolesnici s ovom bolesti dobro reagiraju na terapiju bisfosfonatima (simptomatsko poboljšanje, pad vrijednosti alkalne fosfataze, smanjenje opsega koštanih promjena) te se bolest može držati pod kontrolom i spriječiti njezino daljnje napredovanje (progresija). Kod neliječenih bolesnika prati se progresija koštanih promjena, veća učestalost pojave prijeloma, kao i veći rizik od razvoja osteosarkoma.
LIZOSOMSKE BOLESTI NAKUPLJANJA
Uvod. Lizosomi su stanične organele koje čine tzv. probavni sustav stanice, s obzirom na to da se u njima odvija najveći dio razgradnje složenih molekula na jednostavnije dijelove, što omogućuju različiti enzimi i transportni mehanizmi. Hidrolitički enzimi (glikozidaze, sulfataze, proteaze, esteraze) unutar lizosoma razgrađuju makromolekule na svoje sastavne dijelove (supstrate) koji se potom različitim mehanizmima (pasivni ili aktivni transport) prenose u citoplazmu stanice gdje ulaze u druge, specifične metaboličke puteve.
Definicija. Lizosomske bolesti nakupljanja rijetke su nasljedne bolesti kod kojih nalazimo poremećaj u funkciji nekih od enzima ili transportnih proteina koji sudjeluju u razgradnji neke od makromolekula posljedično čemu se ona pojačano nakuplja unutar lizosoma što dovodi do narušavanja normalne morfologije i funkcije stanice, a u konačnici i cijeloga organa. Do danas je poznao više od 45 poremećaja koji se ubrajaju u ovu skupinu bolesti, a skupna incidencija čini 1 na 7 500 novorođenčadi. Dominantan put nasljeđivanja ovih bolesti je autosomno-recesivni s nekim rijetkim iznimkama (Fabryjeva bolest, Hunterova bolest).
Klasifikacija. Podjela lizosomskih bolesti nakupljanja zasniva se na strukturi supstrata koji se nakuplja unutar lizosoma: mukopolisaharidoze, sfingolipidoze, lipidoze, oligosaharidoze, mukolipidoze, glikogenoze i dr. U daljnjem tekstu bit će opisane najznačajnije bolesti iz ovih skupina: Gaucherova bolest, Fabryjeva bolest, Niemann-Pickova bolest te Pompeova bolest.
GAUCHEROVA BOLEST
Definicija. Gaucherova bolest predstavlja lizosomsku bolest nakupljanja koju karakterizira poremećena funkcija enzima glukocerebrozidaze koji posreduje razgradnju lipida glukocerebrozida s posljedičnim unutarstaničnim nakupljanjem, što se poglavito odvija u stanicama retikuloendotelnoga sustava. Prevalencija ovog poremećaja kreće se između 1:40 000 i 1:110 000.
Etiopatogeneza. Bolest nastaje uslijed mutacije gena za glukocerebrozidazu (GBA) koji se nalazi na prvom kromosomu (1q21), a do danas je opisano više od 300 mutacija navedenoga gena. Bolest se prenosi autosomno-recesivnim putem. Posljedično ovim mutacijama razvija se nedostatak ili smanjena aktivnost glukocerebrozidaze zbog čega dolazi do smanjene razgradnje glukocerebrozida i njegovoga nakupljanja unutar stanica, a najugroženije su stanice retikuloendotelnoga sustava. Glukocerebrozide u najvećoj mjeri nalazimo u sastavu membrana leukocita i eritrocita koji u stanice retikuloendotelnoga sustava dospijevaju tijekom fiziološkoga procesa razgradnje odumrlih leukocita i eritrocita. S obzirom na navedeno, bolest najčešće zahvaća organe i tkiva u kojima nalazimo stanice retikuloendotelnoga sustava: koštana srž (makrofagi), limfni čvorovi (makrofagi), jetra (Kupfferove stanice), slezena (histiociti) te mozak (periadventske stanice). Zahvaćene stanice uslijed nagomilovanja glukocerebrozida poprimaju karakterističan izgled (izgled poput „zgužvanoga svilenog papira“) te se nazivaju Gaucherovim stanicama. Takve stanice pokazuju i određen stupanj pojačane biološke aktivnosti u smislu pojačane produkcije proupalnih citokina koji mogu dovesti do različitih posljedica (povećana koštana reapsorpcija, sklonost malignomima i sl.).
Klinička slika. S obzirom na klinički tijek i manifestacije, Gaucherova bolest ima tri tipa koji se međusobno razlikuju prema zahvaćenosti središnjega živčanog sustava, vremenu kada se bolest pojavljuje kao i naravi kliničkoga tijeka (brzo ili sporoprogresivna).
Tip 1 je najčešći oblik bolesti, a karakteriziran je izostankom zahvaćenosti središnjega živčanog sustava (izostanak neuroloških manifestacija) te kliničkom pojavnošću u bilo kojoj životnoj dobi (najčešće u drugom desetljeću života). Glavne kliničke manifestacije su mu hepatosplenomegalija i koštani poremećaji. Splenomegalija je izražena, uglavnom je bezbolna, a opisani su i slučajevi infarkta slezene što može oponašati akutni abdomen. Hepatomegalija obično nije udružena s poremećajem funkcije, iako transaminaze mogu biti povišene, a rijetki su i slučajevi razvoja ciroze i/ili hepatocelularnoga karcinoma. Koštani poremećaji nastaju uslijed pojačane reapsorpcije koštanoga tkiva i posljedičnih patoloških fraktura. Ostale promjene mogu uključivati razvoj intersticijske fibroze pluća i plućne hipertenzije, pancitopeniju s posljedičnom sklonošću krvarenju i infekcijama, a veća je učestalost i limfoproliferativnih bolesti. Srčano-žilne i bubrežne manifestacije su rjeđe.
Tip 2 je rjeđi oblik bolesti kojega karakterizira zahvaćenost središnjega živčanog sustava uz ostale visceralne manifestacije, progresivnoga je tijeka i većinom završi smrtnim ishodom unutar prve dvije godine života.
Tip 3 ima varijabilnu kliničku sliku, a obično se manifestira u ranom djetinjstvu. Bolest se ispoljava jednakim manifestacijama kao i tip 1 uz umjerene neurološke manifestacije (horizontalni nistagmus, epilepsija), a određena skupina bolesnika ima i karakteristične kardijalne manifestacije u smislu razvoja valvularnih kalcifikata (povezano sa specifičnom mutacijom GBA gena - D409H).
Dijagnoza. Na Gaucherovu bolest treba posumnjati kod bolesnika s nejasnom hepatosplenomegalijom, osobito ako je udružena s koštanim (patološke frakture) i/ili neurološkim manifestacijama. Potvrda bolesti dobiva se na osnovi dokazivanja smanjene aktivnosti enzima glukocerebrozidaze (< 30 %) u leukocitima ili drugim staničnim kulturama (kožni fibroblasti). S obzirom na velik broj mutacija, molekularna dijagnostika (dokazivanje specifične mutacije) nema veće značenje u svakodnevnoj praksi. Dodatne informacije koje podupiru dijagnozu možemo dobiti na temelju nalaza biopsije jetre ili koštane srži (nalaz Gaucherovih stanica), iako oni nisu presudni.
Liječenje može biti specifično i simptomatsko. Specifično liječenje podrazumijeva primjenu rekombinantnih enzima glukocerebrozidaze (imigluceraza, velagluceraza) te lijekova koji smanjuju supstrat bolesti.
Rekombinantni enzimi glukocerebrozidaze primjenjuju se intravenski, a dokazano smanjuju visceralne manifestacije bolesti (organomegalija, pancitopenija, koštane manifestacije), dok nemaju veći učinak na neurološke manifestacije s obzirom na to da enzimi ne prolaze hematoencefalnu barijeru. Liječenje ovim pripravcima treba započeti što ranije, prije razvoja ireverzbilnih promjena na organima te se provodi doživotno. Imigluceraza se može primjenjivati kod trudnica samo ako je to neophodno, dok se velagluceraza ne smije davati trudnicama.
Terapija za smanjenje supstrata bolesti (miglustat) primjenjuje se peroralno i provodi se kao druga linija liječenja Gaucherove bolesti tip I, kada je primjena rekombinantnih enzima nemoguća. Miglustat se ne preporuča u trudnoći i tijekom dojenja, kao ni kod osoba koje planiraju potomstvo.
Simptomatsko liječenje podrazumijeva liječenje bola, srčanih i plućnih komplikacija, neuroloških manifestacija te dijetalnu prehranu. Splenektomija nema veći učinak na klinički tijek bolesti. U terapiji koštanih poremećaja u obzir dolaze različiti ortopedski zahvati.
FABRYJEVA BOLEST
Definicija. Fabryjeva bolest je lizosomska bolest nakupljanja koju karakterizira poremećena funkcija enzima α-galaktozidaze koji posreduje lizosomsku razgradnju lipida koji na kraju molekule imaju šećer α-galatozil (globotriazil-ceramid, galabiozil-ceramid). Prevalencija bolesti u općoj populaciji iznosi oko 1:117 000, dok je u muškoj populaciji veća s obzirom na X-vezani način nasljeđivanja i iznosi oko 1:40 000.
Etiopatogeneza. Radi se o X-vezanoj nasljednoj bolesti kod koje nalazimo mutaciju gena za α-galaktozidazu (GLA gen) koji se nalazi na dugom kraku X-kromosoma (Xq22.1), a do danas je opisano više od 600 vrsta mutacija. Budući da se radi o X-vezanom nasljeđivanju, od bolesti obolijevaju sinovi koji naslijede mutirani gen, dok su kćeri uglavnom prenositelji i vrlo rijetko razviju blaže kliničke oblike. Posljedično mutacijama GLA gena nalazimo smanjenu aktivnost navedenoga enzima, a bolest postaje klinički simptomatska kada aktivnost bude manja od 5 - 10 %. Uslijed smanjene aktivnosti α-glukozidaze izostaje razgradnja lipida koji na kraju molekule imaju šećer α-galatozil, kao što su globotriazil-ceramid i galabiozil-ceramid te se oni pojačano nakupljaju unutar stanica. Promjene su najizraženije u endotelnim stanicama, stanicama glatke muskulature, kardiomiocitima, epitelnim stanicama bubrega te u neuronima. Ako se bolest ne otkrije u djetinjstvu, bolesnici u zreloj životnoj dobi razvijaju bubrežne i srčano-žilne komplikacije koje mogu dovesti i do smrtnoga ishoda (kronična bubrežna bolest, hipertrofična kardiomiopatija, koronarna bolest, valvularne greške).
Klinička slika. Klasična klinička slika koja se razvija kada je aktivnost enzima manja od 1 % manifestira se već u ranom djetinjstvu, a obilježena je pojavom angiokeratoma praćenih hipohidrozom ili anhidrozom, dok se visceralne manifestacije pojavljuju kasnije u životnoj dobi.
Angiokeratomi se opisuju kao tamnocrvene ili tamnoljubičaste kožne lezije u razni kože ili nešto iznad nje, a najčešće su i najgušće raspoređene u području natkoljenica i donjega dijela trbuha. Hipohidroza (smanjeno znojenje) nastaje uslijed zahvaćenosti krvnih žila i živaca kože oko žlijezda znojnica, a posljedično nemogućnosti izdvajanja temperature, bolesnici navode napadaje i navale crvenila i vrućice. Zbog poremećaja perifernih živaca česte su parestezije i bolovi ekstremiteta, a velik dio bolesnika ima i okularne smetnje (zamućenje rožnice).
Ako se bolest ne prepozna, ona postupno progredira i razvijaju se znakovi zahvaćenosti visceralnih organa, u prvom redu srca (hipertrofija lijeve klijetke, koronarna bolest, valvularne greške, a osobito mitralna insuficijencija, bolesti provođenja) i bubrega (postupan razvoj kronične bubrežne bolesti). Ove se manifestacije obično pojavljuju između trećega i petog desetljeća života. Kod bolesnika koji imaju aktivnost enzima između 5 i 10 % bolest se može očitovati samo zahvaćanjem srca i/ili bubrega u odrasloj dobi, što otežava postavljanje dijagnoze.
Ostale kliničke manifestacije javljaju se rjeđe, a uključuju gastrointestinalne smetnje (bolovi u trbuhu, sindrom malapsorpcije), neurološke manifestacije (glavobolje, vrtoglavice, cerebrovaskularni inzult), koštane poremećaje (osteopenija, osteoporoza) i dr.
Dijagnoza. Na dijagnozu treba pomišljati kod djece s angiokeratomima, očnim promjenama, poremećajem znojenja i parestezijama, a u odrasloj dobi kod bolesnika s nerazjašnjenom kardiomiopatijom, renalnom bolesti i trombotskim incidentima. Kod sumnje na ovaj poremećaj potrebna je šira dijagnostička obrada s ciljem utvrđivanja proširenosti bolesti i zahvaćenosti organa: ultrazvuk abdomena, rendgenogram srca i pluća, ehokardiografija, magnetna rezonanca mozga, oftalmološki pregled. Konačna dijagnoza se potvrđuje nalazom smanjene aktivnosti enzima (< 30 %) u leukocitima ili drugim staničnim kulturama (kožni fibroblasti), a moguće je i dokazati specifičnu mutaciju GLA gena molekularnim dijagnostičkim metodama.
Liječenje. Specifično liječenje uključuje primjenu supstitucijskih enzima, a na raspolaganju imamo intravenske pripravke (agalzidaza alfa, agalzidaza beta) te peroralni pripravak (migalastat). Specifično liječenje treba započeti što ranije, prije pojave ireverzibilinih promjena na srcu i bubrezima, te se ono treba primjenjivati doživotno. Simptomatska terapija uključuje liječenje bola, dermatološko uklanjanje angiokeratoma, liječenje bubrežne bolesti, liječenje srčane bolesti te dijetnu ishranu s malo masti.
NIEMANN-PICKOVA BOLEST TIP A I B
Definicija. Niemann-Pickova bolest (tip A i B) predstavlja lizosomsku bolest nakupljanja koju karakterizira poremećena funkcija enzima sfingomijelinaze s posljedičnim intracelularnim nakupljanjem lipida sfingomijelina, primarno u stanicama retikuloendotelnoga sustava. Niemann-Pickova bolest tip C ne pripada ovoj skupini bolesti, nego se radi o poremećaju transporta kolesterola u stanice.
Etiopatogeneza. Tip A i B Niemann-Pickove bolesti predstavljaju autosomno-recesivne nasljedne poremećaje koje karakterizira mutacija SMPD1 gena smještenoga na 11. kromosomu, koji je odgovoran za stvaranje enzima kisele sfingomijelinaze. Posljedično mutacijama toga gena nastaje enzim sa smanjenom aktivnošću uslijed čega izostaje razgradnja sfingomijelina i on se nakuplja unutar stanice i dovodi do karakterističnih promjena. Ako je aktivnost enzima manja od 5 %, razvija se tip A bolesti kojega karakterizira rani i brzi razvoj s obično smrtnim ishodom unutar prve tri života, dok bolesnici s aktivnošću enzima između 5 i 10 % imaju tip B bolesti koji se prezentira u kasnijoj dobi i ima manje progresivan tijek. Bolest pogađa ponajprije tkiva i organe koji sadržavaju veće količine retikuloendotelnih stanica (jetra, slezena, koštana srž, mozak).
Klinička slika. Niemann-Pickova bolest tip A razvija se unutar prva tri mjeseca života te je obilježena brzim i progresivnim tijekom. Kliničkom slikom dominiraju hepatosplenomegalija, neurodegenerativni poremećaji te zastoj psihomotornoga razvoja sa smrtnim ishodom unutar prve tri godine života. Niemann-Pickova bolest tip B ima manje izražen progresivan tijek i obično se dijagnosticira u odrasloj dobi. Obilježena je primarno hepatomegalijom koja kod manjega broja oboljelih može rezultirati razvojem ciroze te plućnim simptomima uslijed smanjenoga difuzijskog kapaciteta i rekurentnih bronhopneumonija. Pojava neuroloških manifestacija je rjeđa pojava (nistagmus, ekstrapiramidni simptomi, kognitivne smetnje). Ostale manifestacije uključuju splenomegaliju, pancitopeniju te kroničnu bubrežnu bolest.
Dijagnoza. Postavljanje dijagnoze ove bolesti otežana je zbog nespecifičnih simptoma i znakova. U dijagnostičkoj obradi u laboratorijskim nalazima obično nalazimo pancitopeniju, umjereno povišene transaminaze te povišene vrijednosti LDL kolesterola; na radiogramu pluća opisuje se retikulonodularni crtež, čak i kod bolesnika koji nemaju plućne simptome, a na spirometriji nalazimo restriktivne smetnje disanja. Konačna se dijagnoza postavlja određivanje aktivnosti kisele sfingomijelinaze u leukocitima ili staničnoj kulturi (kožni fibroblasti). Danas je moguća i molekularna dijagnostika i potvrda mutacije SMPD1 gena (deltaR608 je najčešća mutacija u tipu B). Dijagnozu potpomaže nalaz tzv. Niemann-Pickovih stanica (pjenaste stanice krcate lipidnim sadržajem) u bioptatu koštane srži.
Liječenje. Specifičnoga liječenja nema te se kod bolesnika poduzimaju simptomatske mjere kao što su praćenje i liječenje jetrenoga zatajenja, terapija statinima i dijetna ishrana kod hiperkolesterolemije, primjena krvnih pripravaka kod izražene pancitopenije, oksigenoterapija kod razvoja intersticijske plućne bolest i sl.
POMPEOVA BOLEST
Definicija. Pompeova bolest je lizosomska bolest nakupljanja glikogena koja nastaje kao posljedica poremećene aktivnosti enzima kisele α-glukozidaze. Učestalost bolesti u općoj populaciji iznosi oko 1:40 000 novorođenčadi.
Etiopatogeneza. Radi se o autosomno-recesivno nasljednoj bolesti koja nastaje zbog mutacije GAA gena koji se nalazi na 17. kromosomu (17q25.3), a odgovoran je za stvaranje kisele α-glukozidaze, enzima koji u lizosomima sudjeluje u razgradnji glikogena. Posljedično manjku, odnosno smanjenoj aktivnosti ovoga enzima dolazi do unutarstaničnoga nakupljanja glikogena, a to je najizraženije u mišićima, srcu i plućima.
Klinička slika. Infantilni oblik je obilježen razvojem bolesti u prvih nekoliko mjeseci života, a obilježen je miopatijom, hipotonijom, hepatomegalijom, kardiomegalijom te respiratornom insuficijencijom. Bolest je progresivnoga karaktera i djeca umiru unutar prve godine života. Adultni oblik se javlja kasnije, obično između trećega i petog desetljeća života, a glavna su mu obilježja progresivna slabost proksimalnih mišićnih skupina te slabost respiracijskih mišića. Ako se bolest ne prepozna, napreduje prema potpunoj invalidnosti, razvoju respiratorne insuficijencije i ovisnosti o mehaničkoj ventilaciji. Uz to, prisutna je i kardiomegalija i razvoj kroničnoga srčanog popuštanja.
Dijagnoza. U praksi se dijagnoza adultnoga oblika postavlja relativno kasno, s obzirom na specifičnost kliničkih očitovanje (u pravilu 10 do 15 godina od pojave prvih simptoma). Dijagnostički postupak kod sumnje na ovaj poremećaj uključuje široku neurološku, kardiološku i pulmološku obradu kojom dobivamo uvod u proširenost bolesti i stupanj disfunkcije. Definitivna dijagnoza postavlja se mjerenjem aktivnosti enzima u leukocitima iz periferne krvi ili kulture stanica (fibroblasti) te dokazivanjem mutacija GAA gena molekularnim metodama.
Liječenje. Specifično liječenje uključuje primjenu rekombinantnoga enzima aglukozidaza-α, a liječenje odraslih indicirano je samo ako se radi o simptomatskoj bolesti i značajno sniženoj aktivnosti enzima. Lijek ima smisla primjenjivati prije pojave ireverzibilnih promjena. Uz to se provode klasične mjere simptomatskoga liječenja (liječenje srčanoga popuštanja, liječenje respiratorne insuficijencije, fizikalna terapija i sl.).
BOLESTI NAKUPLJANJA GLIKOGENA (GLIKOGENOZE)
UVODNE NAPOMENE
Glikogen je skladišni oblik glukoze u organizmu, a predstavlja polimer izgrađen od brojnih molekula glukoze. Primarno nastaje u stanicama jetre i skletenih mišića, no može nastati i u nekim drugim stanicama (bubreg, miokard, crijeva). Proces sinteze (glikogeneza) i proces razgradnje (glikogenoliza) omogućavaju brojni enzimi, a defekt bilo kojega od njih može rezultirati ovim poremećajima. Procesi stvaranja i razgradnje glikogena u jetri odgovorni su za regulaciju šećera u krvi (pri gladovanju dolazi do glikogenolize i otpuštanja glukoze u cirkulaciju), dok u skeletnim stanicama osiguravaju izvor energije za mišićni rad (glikogenoliza se odvija s ciljem osiguravanja glukoze potrebne kao izvor energije).
Definicija. Bolesti nakupljanja glikogena (glikogenoze) predstavljaju skupinu rijetkih nasljednih bolesti koje nastaju kao posljedica poremećene sinteze i/ili razgradnje glikogena pri čemu dolazi do njegovoga povećanoga intracelularnog nakupljanja. Najčešće zahvaćeni organi su jetra, skeletni mišići i bubrezi jer su to mjesta dominantnoga stvaranja glikogena. Procijenjena učestalost ovih poremećaja iznosi 1 na 25 000 poroda.
Etiopatogeneza. Radi se o nasljednim bolestima koje se obično prenose autosomno-recesivnim putem, a zahvaćeni geni su oni koji određuju enzime koji sudjeluju u procesu stvaranja ili razgradnje glikogena. Do sada je utvrđeno petnaest različitih poremećaja, a svima je zajedničko pojačano stvaranje ili smanjena razgradnja glikogena pri čemu se on nakuplja unutar stanice, što dovodi do poremećaja morfologije i funkcije organa.
Klinička slika. Budući da su dominantno zahvaćeni jetra i skeletni mišići, osnovni simptomi i znakovi su hepatomegalija i hipoglikemija (odraz zahvaćenosti jetre) te mišićna slabost (odraz zahvaćenosti mišića).
Dijagnoza. Na dijagnozu trebaju upućivati osnovne kliničke manifestacije (hepatomegalija, hipoglikemija i mišićna slabost), a konačna potvrda dobiva se genetskim testiranjem, tj. dokazivanjem specifičnih mutacija na molekuli DNA izoliranoj iz krvi ili sline.
Liječenje. Osnovu liječenja čini osiguravanje stalne nutritivne potpore kako bi se osigurala normoglikemija i izbjegla hipoglikemija uz simptomatske korekcije ostalih nastalih metaboličkih poremećaja koji se mogu javiti. Supstitucijsko liječenje u obzir dolazi kod nekih oblika glikogenoza (npr. Pompeova bolest).
NAJČEŠĆI KLINIČKI OBLICI GLIKOGENOZA
Glikogenoza tip I (von-Gierkeova bolest) nastaje kao posljedica nedostatka enzima glukoza-6-fosfataze, a primarno zahvaćeni organi su jetra i bubreg (hepatorenalna glikogenoza). Klinički se bolest manifestira hepatomegalijom i renomegalijom, a u laboratorijskim nalazima pratimo hipoglikemiju, hiperlipidemiju, hiperuricemiju te metaboličku acidozu. Dijagnoza se postavlja na temelju laboratorijskih nalaza, nalaza biopsije jetre (povećana količina glikogena u hepatocitima, odsustvo fibroze) te molekularnim metodama. Liječenje je simptomatsko: osiguravanje nutritivnoga unosa kako bi se spriječila hiperglikemija uz primjenu ostalih lijekova: alopurinol, hipolipemici, ACE inhibitori (nefroprotekcija).
Glikogenoza tip II (Pompeova bolest) nastaje kao posljedica nedostatka enzima α-glukozidaze koja pripada skupini lizosomalnih hidrolitičkih enzima. Bolest je opisana u poglavlju o lizosomskim bolestima nakupljanja.
Glikogenoza tip III (Corijeva bolest) nastaje kao posljedica nedostatka enzima odgranjavanja (engl. debranching enzyme) koji posreduje razgradnju glikogena u točki grananja. Bolest dominantno pogađa jetru i mišiće, pa se klnički očituje hepatomegalijom i hipoglikemijom koji su slabije izraženi nego u tipu I te miopatijom i hipotonijom. Dijagnoza se postavlja na osnovi kliničke slike, laboratorijskih nalaza, spektrofotometrijskom analizom glikogena u eritrocitima ili bioptatu jetre (kratki bočni lanci) te potvrđuje izravnim mjerenjem aktivnosti enzima i molekularnim metodama (dokazivanje mutacije). Liječenje je simptomatsko (prevencija hipoglikemije, visokoproteinska dijeta).
Glikogenoza tip IV (Andersenova bolest) nastaje kao posljedica nedostatka enzima grananja, enzima koji posreduje nastajanje nove grane glikogena. Kod ovoga deficita dolazi do nakupljanja glikogena abnormalne strukture (neobično dugi lanci s malo grananja). Takav glikogen organizam prepoznaje kao stranu molekulu te pokreće imunološku reakciju. Bolest zahvaća gotovo sve organe i tkiva, očituje se vrlo rano u životu i u pravilu završava smrću unutar nekoliko godina života. Dominantno je zahvaćena jetra uz pojavu ciroze. Liječenje je simptomatsko, opisani su slučajevi transplantacije jetre koja je dovela do produljenja životnoga vijeka.
Glikogenoza tip V (McArdleova bolest) nastaje kao posljedica nedostatka enzima mišićne fosforilaze koji onemogućava iskorištavanje glukoze spremljene u obliku glikogena u mišićima. Glavne kliničke tegobe vezane su uz pojavu bolova u mišićima prilikom jačega mišićnog rada. Dijagnoza se potvrđuje na temelju biopsije mišića koja pokazuje povećanu koncentraciju glikogena normalne strukture unutar miocita. Metabolizam glikogena u jetri je očuvan pa se ne razvijaju hipoglikemije. Liječenje se sastoji od prevencije simptoma uzimanjem veće količine ugljikohidrata neposredno prije fizičkoga napora.
Glikogenoza tip VI (Hersova bolest) nastaje kao posljedica nedostatka enzima jetrene fosforilaze zbog čega je poremećena razgradnja glikogena samo u jetri s posljedičnim nakupljanjem (hepatomegalija) i pojavom hipoglikemije natašte. Dijagnoza se postavlja na temelju glukagonskoga testa koji podrazumijeva primjenu glukagona koji potiče jetrenu fosforilazu te se normalno očekuje porast glukoze u krvi nakon davanja, što pri ovome poremećaju izostaje. Dijagnozu potvrđujemo mjerenjem aktivnosti enzima i molekularnim metodama. Liječenje se sastoji od nutritivne potpore kojom se sprečava razvoj hipoglikemije.
POREMEĆAJI PREHRANE: PRETILOST, METABOLIČKI SINDROM I MALNUTRICIJA
Pretilost i metabolički sindrom dva su usko povezana poremećaja koji danas predstavljaju jedan od najvećih javnozdravstvenih problema razvijenih zemalja jer predstavljaju jedan od ključnih rizičnih čimbenika u razvoju najčešćih oboljenja, kao što su srčanožilne bolesti, neoplazme, degenerativne bolesti lokomotornoga sustava i druge. S druge strane, proteinsko-energetska malnutricija više se veže uz nerazvijene zemlje, osobito uz djecu, dok je u razvijenim zemljama češća kod starijih osoba kao posljedica kroničnih, iscrpljujućih bolesti i stanja.
PRETILOST
Definicija. Pretilost (debljina, adipozitet) definira se kao stanje koje karakterizira pojačano nakupljanje masnoga tkiva u organizmu, a dovodi do neželjenih posljedica po zdravlje pojedinaca. Pretilost se najčešće izražava indeksom tjelesne mase (BMI, od engl. body mass index) – Tablica 8.33.
|
Tablica 8.33. Definicija pretilosti izražena indeksom tjelesne mase
|
|
BMI < 18,5
|
Premala tjelesna težina
|
|
BMI 18,5 - 24,9
|
Normalna tjelesna težina
|
|
BMI 25,0 - 29,9
|
Prekomjerna tjelesna težina
|
|
BMI 30,0 - 34,9
|
Debljina I. stupnja
|
|
BMI 35,0 - 39,9
|
Debljina II. stupnja
|
|
BMI > 40
|
Debljina III. stupnja, morbidna debljina
|
Epidemiologija. Nedvojbeno je da je pretilost danas jedan od najznačajnijih javnozdravstvenih problema, s obzirom da se smatra da više od 2 milijarde ljudi ima prekomjernu tjelesnu težinu, a više od 700 milijuna je pretilo. Ovo je izraženije u razvijenim zemljama i zemljama u razvoju, što je uvjetovano promjenom životnoga stila i navika (sjedilački način života). Također, bilježi se i sve veći postotak pretile djece, kao i pojavnost pretilosti u starijoj životnoj dobi. Nešto viša prevalencija pretilosti bilježi se kod žena u odnosu na muškarce. Prema podacima Svjetske zdravstvene organizacije pojavnost (prevalencija) debljine u svijetu gotovo se utrostručila od 1975. do 2016. godine. U Republici Hrvatskoj prekomjernu tjelesnu masu i debljinu ima 57,4 % osoba starijih od 18 godina, od čega je s prekomjernom tjelesnom masom 38,7 % stanovnika, a debelih je 18,7 %. U Republici Hrvatskoj prekomjerna tjelesna masa i debljina prisutne su kod 34,9 % djece, izraženije kod dječaka u odnosu na djevojčice.
Etiopatogeneza. Pretilost nastaje kao posljedica skladištenja viška energije u organizmu u obliku masti, odnosno masnoga tkiva. Taj višak nastaje kao posljedica povećanoga unosa energije putem hrane i/ili njezinoga smanjenog iskorištavanja (tjelesna neaktivnost). Pretilost se razvija kao posljedica interakcije više čimbenika koji uključuju genetsku osnovu, vanjske čimbenike i ponašanje, kao i pridružene bolesti (Tablica 8.34.). Nagao porast prevalencije debljine diljem svijeta uvjetovan sjedilačkim načinom života i promjenama prehrambenih navika, kao i tjelesne aktivnosti, u prvi plan u razvoju pretilosti stavlja vanjske čimbenike i ponašanje pojedinaca i ukazuje na njihovu važnost.
Genetska osnova pretilosti. Danas se zna da postoji više gena koji mogu biti povezani s nastankom debljine, a svi su oni uključeni u fiziološki put osjeta gladi kojega čine periferne masne stanice i centar za glad koji se nalazi u hipotalamusu. U stanju pojačanoga unosa hrane i pojačane pohrane masti u masnim stanicama one luče hormon leptin koji djeluje na neurone nukleusa arkuatusa koji potom stvara tzv. anoreksigene peptide koji djeluju na hipotalamus tako da smanjuju osjet gladi (POMC - proopiomelanokortin, CRH - kortikoliberin, TSH - tireoliberin). Također, u ovim neuronima stvaraju se i tzv. oreksigeni peptidi koji potiču osjet gladi u stanjima smanjene koncentracije leptina u krvi (NPY - neuropeptid Y, MCH - melanin concentrating hormone). Osim leptina, sličan učinak imaju i drugi peptidi, kao što su adiponektin, grelin, inkretini, interleukini 1 i 6 i dr. Samo se mali broj slučajeva pretilosti može objasniti mutacijom jednoga gena (obično gena za leptin ili leptinski receptor), a uglavnom se radi o interakciji poremećaja više gena zajedno s vanjskim čimbenicima (ponašanje). Pretilost se može naći i kao dio nekih od nasljednih genetskih sindroma: Prader-Willi sindrom, Laurence-Moon-Biedl sindrom, Ahlstromov sindrom, Cohenov sindrom i drugi.
Vanjski čimbenici i ponašanje pojedinaca. Ishrana i fizička aktivnost dva su glavna čimbenika koji imaju ulogu u nastanku pretilosti. Prekomjeran unos rafiniranih ugljikohidrata i masti (osobito životinjskoga porijekla) danas su najveći krivci za razvoj pretilosti. Tjelesna neaktivnost također je jasno povezana s nastankom pretilosti i drugi je najveći uzrok epidemije debljine. Smanjena tjelesna aktivnost dovodi do smanjenja iskorištavanja energije pohranjene u masnom tkivu te tako pogoduje nastanku pretilosti. Određeni obrasci ponašanja kao što je kompulzivno prejedanje (naglo unošenje veće količine hrane u kraćem vremenskom razdoblju) također pridonose nastanku pretilosti.
Pridružene bolesti i stanja. Pretilost se može razviti i kao posljedica određenih bolesti i stanja: Cushingov sindrom, hipotireoidizam, inzulinom te bolesti hipotalamusa (trauma, tumori, upale). Nastanku pretilosti pridonose i neki drugi čimbenici, kao što su manjak sna, primjena psihotropnih lijekova (starije generacije anksiolitika i antidepresiva), prestanak pušenja i sl.
|
Tablica 8.34. Uzroci pretilosti
|
|
Primarni uzroci pretilosti
|
Sekundarni uzroci pretilosti
|
|
Monogenetski poremećaji: mutacija melanokortin-4 receptora, deficijencija leptina, deficijencija POMC;
Genetski sindromi: Prader-Willi, Bardet-Biedl, Cohen, Alstrom, Froehlich.
|
Neurološki: ozljede i tumori mozga, hipotalamička pretilost;
Endokrini: hipotireoza, Cushingov sindrom, deficijencija hormona rasta, pseudohipoparatireoidizam;
Psihološki: depresija, poremećaji prehrane (unosa hrane);
Lijekovi: triciklički antidepresivi, oralni kontraceptivi, antipsihotici, antikonvulzivi, glukokortikoidi, preparati sulfonilureje, glitazoni, beta blokatori.
|
Klinička slika. Pretilost se u prvom planu klinički manifestira povećanjem tjelesne mase, a potom i izravnim i neizravnim simptomima i znakovima svojih posljedica. Debljina se karakterizira prema veličini i broju masnih stanica. Hipertrofična je ona u kojoj je povećana veličina masnih stanica, dok je hiperplastična ona u kojoj je povećan broj masnih stanica. Broj masnih stanica može se povećati samo u doba razvoja (intrauterino, do prve tri godine života i u doba puberteta). Izrazito debele osobe imaju obično miješani tip debljine (hipertrofični i hiperplastični). U svojoj osnovi, a prema rasporedu nakupljanja masnoga tkiva razlikujemo dva tipa pretilosti: centralni (abdominalni ili visceralni) - pri kojemu se masno tkivo nakuplja unutar trbušne šupljine te periferni (potkožni) - pri kojemu se masno tkivo nakuplja poglavito u potkožju. Abdominalni tip pretilosti povezan je s poremećajem izlučivanja hormona masnoga tkiva i metaboličkim promjenama s posljedičnim komplikacijama. Uzrokuje kroničnu upalu koja podržava nastanak inzulinske rezistencije te posljedično pojavu metaboličkoga sindroma. Također, raspored masnoga tkiva uvjetovan je i spolom, pa se kod žena mast tipično nakuplja u predjelu bokova i zdjelice (kruškoliki oblik), dok se kod muškaraca nakuplja u području struka i gornjega dijela trbuha (jabukoliki oblik).
Dijagnoza. U procjeni debljine služimo se različitim načinima: izračunavanje indeksa tjelesne mase, mjerenjem opsega struka, izračunavanjem omjera struk/kuk, mjerenjem kožnih nabora te posebnim tehnikama određivanja postotka tjelesne masti.
Indeks tjelesne mase (BMI, body mass index) najčešći je korišteni alat za procjenu debljine. Računa se matematički tako da se podijeli tjelesna masa u kilogramima sa kvadratom visine u metrima. Prema dobivenim vrijednostima, osobe svrstavamo u jednu od šest kategorija (Tablica 8.33.).
Opseg struka također se često primjenjuje u procjeni pretilosti. Mjerenje se vrši centimetarskom vrpcom koja se postavlja oko struka na polovini udaljenosti između donjega ruba rebrenoga luka i kriste ilijake bočno i to za vrijeme ekspirija. Patološki nalaz je opseg struka veći od 94 cm za muškarce i 80 cm za žene.
Omjer struk/kuk (WHR, od engl. waist to hip ratio) dobiva se mjerenjem opsega struka na najuužem dijelu i opsega bokova u najširem dijelu te dijeljenjem tih vrijednosti (struk/kuk). Patološkim se smatra ako je veći od 0,75 - 0,85 za žene i 0,8 - 1,0 za muškarce.
Određivanje postotka tjelesne masti može se vršiti na nekoliko načina: tehnika podvodnoga vaganja, bioelektrična impedancija, denzitometrija te CT ili MR snimanjem što je izuzetno rijetko. Najčešće se koristi bioelektrična impendancija koja vrši izračun postotka masnog tkiva na osnovi prolaska električne struje niske energije kroz tijelo, a pouzdano definira volumen masnoga tkiva. Normalan postotak masnoga tkiva kod muškaraca je 10 do 20 %, a kod žena 20 do 30 %.
U dijagnostičkoj obradi pretilih osoba treba obuhvatiti i misliti na moguće bolesti i stanja koja su dovela do debljine te na bolesti i stanja koja mogu proizaći iz debljine (hiperlipidermija, arterijska hipretnzija, hiperglikemija i dr.). U tu svrhu potrebno je odrediti hormone štitnjače, isključiti hiperkorticizam kod onih koji imaju kliničke znakove koji na njega upućuju, kod muškaraca sa simptomima i znakovima hipogonadizma odrediti LH, FSH, ukupni testosteron, SHBG (globulin koji veže spolne hormone), a kod žena s neregularnim ciklusima te sterilitetom odrediti LH, FSH, ukupni testosteron, SHBG, androstendion, estradiol, 17-hidroksiprogesteron i proklacitoin (primarno radi isključenja sindroma policističnih jajnika).
Posljedice pretilosti. Danas je jasno da masno tkivo ne djeluje samo kao skladište energije i termoizolator, nego je ono posebni endokrini organ koji stvara i luči različite hormone i aktivne peptide (adipokini) zbog čega je debljina povezana s pojavom različitih bolesti i stanja: šećerna bolest tip II, arterijska hipertenzija, hiperlipidemija, kardiovaskularne bolesti, cerebrovaskularne bolesti, gastrointestinalne bolesti, degenerativne bolesti lokomotornoga sustava (osteoartritis), ginekološki i reprodukcijski problemi, endokrinološki poremećaji, maligni tumori, opstruktivna apenija u spavanju te psihološki problemi. Već i smanjenje tjelesne mase od samo 5 do 10 % od početne tjelesne mase smanjuje rizik obolijevanja od niza bolesti povezanih s debljinom.
Liječenje pretilosti u prvom redu podrazumijeva pravilnu ishranu i povećanje tjelesne aktivnosti, dok su farmakološko i kirurško liječenje ostavljeni za teže oblike. U Tablici 8.35. navedene su preporuke za liječenje pretilosti (National Heart, Lung and Blood Institute, North American Association for the Study of Obesity).
|
Tablica 8.35. Preporuke za liječenje pretilosti
|
|
|
BMI 25,0 - 26,9
|
BMI 27,0 - 29,9
|
BMI 30,0 - 34,9
|
BMI 35,0 - 39,9
|
BMI > 40
|
|
Prehrana, tjelovježba
|
Uz komorbiditet
|
Uz komorbiditet
|
Bez obzira na komorbiditet
|
Bez obzira na komorbiditet
|
Bez obzira na komorbiditet
|
|
Primjena lijekova
|
Ne
|
Uz komorbiditet
|
Bez obzira na komorbiditet
|
Bez obzira na komorbiditet
|
Bez obzira na komorbiditet
|
|
Kirurško liječenje
|
Ne
|
Ne
|
Ne
|
Uz komorbiditet
|
Bez obzira na komorbiditet
|
Prehrana i tjelesna aktivnost. Restrikcija kalorijskoga unosa (dijeta) i pojačanje tjelesne aktivnosti osnovne su mjere liječenja pretilosti. Do sada se nijedna dijeta koja je uključivala apsolutno isključenje određenih namirnica ili njihovo cikličko uzimanje nije pokazala učinkovitom i povoljnom. Bolesnici moraju uzimati raznoliku hranu uz povećanje unosa voća i povrća i smanjenje unosa rafiniranih ugljikohidrata i masti. Energetski unos trebao bi se smanjiti za oko 500 do 1000 kCal od onoga što su uzimali prije, čime se osigurava postupan gubitak tjelesne mase za oko 0,45 do 0,90 kg na tjedan. Preporuča se uzimati više manjih obroka tijekom dana te izbjegavati unos hrane pred spavanje. Uz prehranu osobe moraju intenzivirati svoju svakodnevnu tjelesnu aktivnost (više hodanja, izbjegavanje korištenja lifta i sl.) uz dodatak umjerene fizičke aktivnost u trajanju od 30 minuta na dan (brz hod, tjelovježba i sl). Ne preporuča se naglo mršavljenje s obzirom na to da se u masnom tkivu nalaze otopljeni brojni toksini, zbog čega se oni naglo otpuštaju u krv i dovode do toksemije.
Farmakoterapija pretilosti. Primjena lijekova kod pretilih osoba čini nadopunu liječenja prehranom i tjelesnom aktivnošću i nije im zamjena. Na raspolaganju je nekoliko lijekova iz različitih skupina: orlistat, lorkaserin, fentermin/topiramat, naltekson/bupropion te liraglutid. U Republici Hrvatskoj za liječenje pretilosti registrirani su naltrekson/bupropion, orlistat i liraglutid.
Orlistat djeluje u probavnom sustavu inhibirajući crijevnu lipazu čime dovodi do smanjene apsorpcije unesenih masti. Primjenjuje se u dozi od 120 mg do tri puta na dan uz obroke koji sadrže veće količine masti. Glavne nuspojave su lokalne prirode, na primjer, masne proljevaste stolice, grčevi i flatulencija, a može dovesti i do malapsorpcije vitamina topljivih u mastima (D, E, K, A). Može se koristiti kao dugoročna farmakoterapija pretilosti ako se dobro podnosi.
Lorkaserin djeluje kao selektivni agonist serotininskih receptora čime se potiče stvaranje POMC-a, jednoga od snažnih peptida koji djeluju na hipotalamus izazivajući smanjenje osjeta gladi. Koristi se peroralno u dozi od 10 mg dva puta na dan. Nastanak valvularne srčane bolesti povezane s primjenom neselektivnih agonista serotoninskih receptora (fenfluramin, deksfenfluramin) nije dokazan kod primjene lorkaserina.
Fentermin/topiramat je lijek koji se koristi u liječenju pretilosti, a sadrži kombinaciju fentermina (anoreksik) te topiramata (antikonvulziv). Fentermin djeluje slično kao i amfetamini s kojima i pokazuje određene kemijske sličnosti i smanjuje osjet gladi, a za topiramat su kliničke studije koje su provođene kod bolesnika s epilepsijom i migrenom pokazale da značajno smanjuje tjelesnu težinu. Primjenjuje se u dozi od 3,75/23 mg prvih 14 dana, a potom 7,5/46 mg do maksimalno 15/92 mg jednom na dan. Doziranje lijeka vrši se prema učinku (ako se s osnovnom dozom ne postigne gubitak tjelesne mase nakon 14 dana uzimanja lijeka, može se preći na veću dozu). Najčešće nuspojave su promjene raspoloženja, umor i nesanica. Dokazano je da ovaj lijek dovodi i do ubrzanja srčane frekvencije pa nije jasna povezanost s kardiovaskularnim rizikom. Također, lijek dokazano povećava učestalost anomalija ploda ako se uzima za vrijeme trudnoće.
Naltekson/bupropion predstavlja kombinaciju antagonista opioidnih receptora (naltrekson) te inhibitora ponovne pohrane dopamina i norepinefrina (bupropion). Obje komponente lijeka učinak postižu djelujući na hipotalamus smanjujući osjet gladi. Početna doza iznosi 8/90 mg jednom na dan s postupnim povećanjem doze od 1 tablete svaki tjedan do maksimalne doze od dvije tablete dva puta na dan. Opisivane nuspojave uključuju poremećaje raspoloženje, pojavu suicidalnih misli, glavobolju, mučninu i povraćanje, a dovode i do porasta krvnoga tlaka i srčane frekvencije.
Liraglutid je inkretinski analog GLP-1 (glucagon-like peptide-1) koji je inicijalno odobren za liječenje šećerne bolesti tip II. Kako GLP-1 djeluje i na hipotlamus smanjujući osjet gladi , a usporava i praženjenje želuca i produžava osjećaj sitosti. Primjenjuje se u obliku supkutane injekcije najprije u dozi od 0,6 mg povećavajući je za 0,6 mg tjedno do maksimalne doze od 3 mg/dan. Uobičajene nuspojave ovoga lijeka su mučnina, povraćanje, proljev, konstipacija i hipoglikemija koje su uglavnom prolazne, a potreban je oprez pri primjeni kod bolesnika s kolelitijazom i pankreatitisom.
Kirurško liječenje pretilosti rezervirano je za najteže oblike pretilosti. Posebna grana kirurgije koja se bavi kirurškim liječenjem debljine naziva se barijatrijska kirurgija. Trenutni kirurški zahvati u području barijatrijske kirurgije mogu se podijeliti u tri skupine ovisno o mehanizmu kojime dovode do gubitka tjelesne mase: restriktivni (smanjuju želučani kapacitet i sposobnost pohrane hrane u njemu), malapsorptivni (različite crijevne premosnice kojima se isključuju dijelovi tankoga crijeva iz probavne cijevi) te njihove kombinacije. Najčešće operacije u barijatrijskoj kirurgiji su želučani prsteni (engl. gastric banding), želučane premosnice, sleeve gastrekotmija te biliopankreasna diverzija s duodenalnim prekapčanjem (engl. switch).
Dijagnostička obrada, liječenje i praćenje bolesnika s pretilosti, a osobito ako se radi o morbidnoj pretilosti, zahtijeva multidisplinaran pristup u kojemu sudjeluju endokrinolozi, gastroenterolozi, kirurzi, nutricionisti te psihoterapeuti. Danas se u svim većim centrima osnivaju ambulante i odjeli za liječenje pretilosti.
Prevencija pretilosti predstavlja javnozdravstveni imperativ današnjice. S programima prevencije treba započeti što ranije, još kroz vrtićku i školsku dob kada se formiraju dugoročne prehrambene navike i oblici ponašanja (primarna prevencija pretilosti). Sekundarna prevencija pretilosti usmjerena je na rizičnoga pojedinca ili rizičnu skupinu s ciljem uočavanja prekomjerne tjelesne težine te njezine povezanosti s drugim komplikacijama. Ovo podrazumijeva edukaciju o pravilnoj prehrani i važnosti tjelesne aktivnosti, kao i redovne kontrole krvnoga tlaka, glikemije, lipidograma te hepatograma. Najveći nositelji prevencije pretilosti trebali bi biti liječnici primarne zdravstvene zaštite, kao i nadležni epidemiološki zavodi. Snažne javnozdravstvene kampanje protiv pretilosti u nekim zemljama već polučuju rezultat kroz polagano smanjenje porasta broja pretilih osoba.
METABOLIČKI SINDROM
Definicija. Metabolički sindrom (sindrom X) definira se kao kliničko stanje koje karakterizira istodobna prisutnost više rizičnih čimbenika koji su međusobno povezani, a povećavaju rizik od nastanka srčano-žilnih bolesti te šećerne bolesti tip 2. O metaboličkom sindromu govorimo ako su kod bolesnika prisutna najmanje tri od sljedećih pet rizičnih čimbenika: abdominalni tip pretilosti, hiperglikemija, arterijska hipertenzija, povišene vrijednosti triglicerida te smanjene vrijednosti HDL kolesterola.
Epidemiologija. Kao i pretilost, metabolički sindrom pokazuje epidemijske razmjere u svojoj pojavnosti u razvijenim zemljama i zemljama u razvoju. Smatra se da udio od jedne petine do jedne četvrtine stanovništva u tim zemljama ima metabolički sindrom, što ponajprije ovisi o definiciji metaboličkoga sindroma (različiti kriteriji za dijagnozu metaboličkoga sindroma).
Etiopatogeneza. Na metabolički sindrom treba gledati kao na skup različitih rizičnih čimbenika koji su međusobno patofiziološki povezani pri čemu je gotovo nemoguće odrediti koji je to ishodišni događaj koji pokreće cijelu patofiziološku priču. Osnovu cijeloj priči daje pretilost i povećana količina masnoga tkiva koje luči različite medijatore koji dovode do različitih patofizioloških učinaka: inzulinske rezistencije, hiperlipidemije, endotelne disfunkcije, arterijske hipertenzije te hiperkoagulabilnosti.
Visceralni tip pretilosti dobro je definiran rizični čimbenik za pojavnost kardiovaskularnih komplikacija. Za razliku od supkutanoga masnog tkiva njegove adipocite karakterizira niska osjetljivost na inzulin, razvijena i dobro prokrvljena stroma te izražena infiltracija upalnim stanicama. Osim toga, visceralno masno tkivo karakterizira izražena endokrinološka aktivnost u vidu stvaranja i lučenja različitih medijatorima koje jednim imenom nazivamo adipokini. Povećana je sinteza proupalnih citokina (TNF-alfa, IL-1, IL-6, PAI-1), dok je snižena sinteza protuupalnih citokina (adiponektin), zbog čega se pretilost smatra stanjem kronične upale niskoga intenziteta (engl. low grade inflammation). Posljedično tome kod ovih bolesnika nalazimo povišene vrijednosti C-reaktivnoga proteina.
Inzulinska rezistencija označava stanje neosjetljivosti stanica na djelovanje inzulina što dovodi do pojačanoga lučenja inzulina i hiperinzulinemije (kompenzacijski mehanizam) kako bi se održalo euglikemijsko stanje. Vežući se za stanični receptor, inzulin dovodi do aktivacije dva signalna puta: jedan je bitan za metaboličke učinke inzulina, vazodilataciju i protuupalni učinak (PI3P, fosfatidil-inozitol 3-kinaza), a drugi je bitan za mitogeni učinak inzulina, odnosno rast i diferencijaciju stanica (MAPK, mitogen-aktivirajuća protein kinaza). U inzulinskoj rezistenciji dominira poremećaj prvoga signalnog puta što rezultira smanjenim unosom glukoze u stanice i razvojem hiperglikemije, kao i smanjenom produkcijom dušičnoga oksida zbog čega se razvija vazokonstrikcija i endotelna disfunkcija. Nastanak inzulinske rezistencije nije do kraja razjašnjen, ali se najviše dovodi u vezu s djelovanjem proupalnih citokina koji remete normalne signalne puteve, a pretilost je stanje kronične upale niskoga intenziteta.
Hiperlipidemija kod ovih bolesnika može nastati uslijed inzulinske rezistencije posljedično čemu se javlja pojačana aktivnost lipoprotein lipaze. Osim toga, adipociti su i prirodno manje osjetljivi na djelovanje inzulina, što pogoduje lipolizi. Povećana lipoliza u masnom tkivu povećava koncentraciju slobodnih masnih kiselina u krvi koje u jetri služe kao supstrat za sintezu triglicerida u obliku VLDL čestica. Trigliceridi iz VLDL čestica mogu se prebaciti u HDL čestice pri čemu nastaju HDL čestice bogate trigliceridima koje se kao takve pojačano uklanjaju putem jetre zbog čega se smanjuje njihova ukupna količina. Povišene vrijednosti triglicerida uz snižene vrijednosti HDL kolesterola dobro su poznati rizični čimbenici u razvoju ateroskleroze i srčano-žilnih bolesti.
Endotelna disfunkcija posljedica je pojačane produkcije vazokonstriktivnih tvari u masnom tkivu te djelovanja prouplanih citokina koji induciraju infiltraciju upalnih stanica u stijenku krvne žile. Osim toga, neki citokini, kao što su TNF-alfa i rezistin iz masnoga tkiva, mogu djelovati endokrino ili parakrino na endotel te izazvati apoptozu endotelnih stanica. Manjak adiponektina u pretilosti također pogoduje endotelnoj disfunkciji s obzirom na to da on u fiziološkim uvjetima djeluje kao stimulator endotelne NO-sintetaze.
Arterijska hipertenzija u metaboličkom sindromu povezana je s više čimbenika. Razvoj endotelne disfunkcije koja dovodi do vazokonstrikcije jedan je od ključnih mehanizama, ali veliku ulogu imaju i pojačana aktivnost simpatikusa koju nalazimo u pretilosti, kao i povećana aktivnost renin-angiotenzin-aldosteron sustava (RAAS). Jedna od endokrinoloških karakteristika masnoga tkiva je i ta da imaju lokalno razvijen RAAS sustav te imaju sposobnost stvaranja angiotenzinogena, ACE enzima te angiotenzinskih receptora. Osim lokalnoga učinka na protok krvi kroz masno tkivo, oni mogu imati i sistemski učinak što pridonosi razvoju arterijske hipertenzije.
Hiperkoagulabilno stanje nastaje interakcijom ranije opisanih događaja. Endotelna disfunkcija i njegovo oštećenje povezani su s povišenim razinama prokoagulantnih faktora u krvi, kao što su von Willenbrandov faktor (vWF), tkivni faktor (TF), faktor VII (FVII) te fibrinogen. Navedeni faktori mogu porijeklom biti iz endotelnih stanica, ali većinu njih sintetiziraju i adipociti. Adipokini djeluju i na trombocite tako da ih aktiviraju. Pojačana aktivacija trombocita zajedno s povećanom koncentracijom prokoagulantih faktora povećava rizik od nastanka trombeombolijskih incidenata kod ovih bolesnika.
Klinička slika. Metabolički sindrom, kao i sama pretilost, predstavljaju asimptomatska stanja koja pogoduju nastanku nekih od komplikacija. Kod bolesnika s metaboličkim sindromom pojavnost kardiovaskualrnih događaja povećana je za pet puta, dok je pojavnost šećerne bolesti tip 2 dvostruko veća u odnosu na opću populaciju. Osim toga, metabolički sindrom povezan je i s češćom pojavom masne promjene jetre, hepatocelularnoga karcinoma i kolangiokarcinoma, kroničnom bolesti bubrega, opstruktivnom apnejom u spavanju, sindromom policističnih jajnika, hiperuricemijom i gihtom.
Dijagnoza metaboličkoga sindroma postavlja se na temelju anamneze, fizikalnoga pregleda te laboratorijskih nalaza u kojima pronalazimo zadane kriterije. Kriteriji za postavljanje dijagnoze metaboličkoga sindroma prikazani su u Tablici 8.36. Da bi govorili o metaboličkom sindromu, bolesnik mora imati minimalno tri od pet navedenih kriterija.
|
Tablica 8.36. Dijagnostički kriteriji za metabolički sindrom
|
|
Kriterij
|
Vrijednost
|
|
Opseg struka
|
≥ 102 cm (muškarci)
≥ 88 (žene)
|
|
Glukoza natašte
|
≥ 5,5 mmol/L
Ili uzimanje antidijabetika
|
|
Krvni tlak
|
≥ 130/85 mmHG
Ili uzimanje antihipertenziva
|
|
Trigliceridi
|
≥ 1,7 mmol/L
Ili uzimanje hipolipemika
|
|
HDL kolesterol
|
≤ 1,03 (muškarci) ili ≤ 1,3 (žene)
Ili uzimanje lijekova za povišenje HDL-a
|
Liječenje. Osnovu liječenja metaboličkoga sindroma kao i kod debljine čini promjena životnih navika u vidu pravilne prehrane i tjelesne aktivnosti. Liječenje ostalih komponenata metaboličkoga sindroma (hiperglikemija, hiperlipidemija, arterijska hipertenzija) provodi se prema standardnim načelima opisanim u odgovarajućim dijelovima udžbenika. Budući da je metabolički sindrom prokoagulabilno stanje, savjetuje se i redovna primjena acetilsalicilne kiseline ako nije kontraindicirana.
PROTEINSKO-ENERGETSKA MALNUTRICIJA
Definicija. Proteinsko-energetska malnutricija je naziv za kliničko stanje koje nastaje kao posljedica nedostatka odgovarajućega unosa svih makro- i mikronutrijenata. Epidemiološki gledano, u nerazvijenim zemljama ovaj poremećaj se uglavnom susreće kod djece uslijed gladovanja, a u razvijenim zemljama se obično susreće kod starijih osoba koje imaju brojne kronične komorbiditete.
Etiopatogeneza. S obzirom na etiologiju, proteinsko-energetska malnutricija može biti primarna i sekundarna. Primarna nastaje kao posljedica nedostatnoga uzimanja hrane (namjerno izgladnjivanje ili nenamjerno gladovanje), dok je sekundarna posljedica različitih bolesti i stanja koja remete normalnu apsorpciju i utilizaciju hranjivih tvari (bolesti probavne cijevi i pridruženih probavnih organa, kronične upalne bolesti, kronične nezarazne bolesti, endokrinopatije). Kao posljedica manjka energije najprije se razgrađuje masno tkivo, a potom i tjelesni proteini, zbog čega bolesnici gube težinu, mršave i postaju kahektični.
Klinička slika. Osnovni klinički znak malnutricije je gubitak tjelesne težine i mršavljenje bolesnika. Uz to se obično javljaju opći simptomi kao što je umor, malaksalost, gubitak snage te apatija i adinamija. Također posljedično manjku određenih mikro- i makronutrijenata, javljaju se karakteristične posljedice kao što su edemi, anemija, petehije, usporeno cijeljene rana, ispadanje kose i sl. Kod najtežih bolesnika razvija se opća malaksalost, bolesnici ostaju nepokretni te dolazi do postupnoga gašenja svih fizioloških funkcija.
Dijagnoza se postavlja na temelju anamnestičkih podataka, kliničkoga nalaza te osnovnih laboratorijskih nalaza s određivanjem specifičnih biokemijskih markera (proteini, albumini, vitamini, manezij, fosfor i sl.). Prema težini kliničke slike, indeksu tjelesne mase, određenim laboratorijskim parametrima (albumini, transferin, limfociti) te indeksu kasne preosjetljivosti, razlikujemo tri stupnja proteinsko-energetske malnutricije (Tablica 8.37.). Indeks kasne preosjetljivosti podrazumijeva otpornost iskazanu prilikom kožnoga testiranja uobičajenim antigenima (Candida spp, Trichophyton spp). Indeks se označava brojevima 0, 1 ili 2 ovisno o promjeru indurata kože (0 = < 0,5 cm, 1 = 0,5 – 0,9 cm, 2 = = 1,0 cm).
|
Tablica 8.37. Kliničko stupnjevanje proteinsko-energetske malnutricije
|
|
Parametar
|
Blaga malnutricija
|
Umjerena malnutricija
|
Teška malnutricija
|
|
Normalna težina ( %)
|
85 - 90
|
75 - 85
|
< 75
|
|
Indeks tjelesne mase
|
18,0 - 18,9
|
16,0 - 17,9
|
< 16
|
|
Albumin (g/dL)
|
3,1 - 3,4
|
2,4 - 3,0
|
< 2,4
|
|
Transferin (mg/dL)
|
201 - 219
|
150 - 200
|
< 150
|
|
Limfociti (na mm3)
|
1501 - 1999
|
800 - 150
|
< 800
|
|
Indeks kasne preosjetljivosti
|
2
|
1
|
0
|
Liječenje je usmjereno na liječenje osnovne bolesti i ispravljanje osnovnoga poremećaja. Bolesnicima je potrebno postupno supstituirati nutrijente koji su deficitarni, bilo peroralni putem, bilo parenteralnom prehranom u teškim oblicima. Naglasak u liječenju ovih bolesnika je na postupnom uvođenju hranjenja i opterećenju nutrijentima kako bi se izbjegle komplikacije kao što su preopterećenje tekućinom, sindrom ponovnoga hranjenja, hiperglikemija i proljev.
- Aganović I., Metelko Ž. Šećerna bolest. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008. str. 1244-64.
- American Diabetes Association. Standards of medical care in diabetes—2018. Diabetes Care. 2018 Jan;41(Suppl):S1–159.
- Bacon BR. Iron overload. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1418-23.
- Barić I. Poremećaji metabolizma aminokiselina. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008. str. 1313-6.
- Blumer I, Hadar E, Hadden DR, Jovanovič L, Mestman JH, Murad MH et al. Diabetes and pregnancy: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2013 Nov;98(11):4227–49.
- Bodamer OA. Approach to inborn errors of metabolism. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1384-9.
- Božikov V., Aganović I. Pretilost i metabolički sindrom. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008. str. 1267-75.
- Božikov V., Čabrijan T. Hipoglikemija. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008. str. 1264-7.
- Cannon CP, Blazing MA, Giugliano RP, McCagg A, White JA, Theroux P et al; IMPROVE-IT Investigators. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med. 2015 Jun 18;372(25):2387–97.
- Chin JA, Sumpio BE. Diabetes mellitus and peripheral vascular disease: diagnosis and management. Clin Podiatr Med Surg. 2014 Jan; 31(1):11–26.
- Crandall J, Shamoon H. Diabetes mellitus. U: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1527-47.
- DeFronzo R, Fleming GA, Chen K, Bicsak TA. Metformin-associated lactic acidosis: current perspectives on causes and risk. Metabolism. 2016 Feb; 65(2):20–9.
- Dietz WH. Obesity and excessive weight gain in young adults: new targets for prevention. JAMA. 2017 Jul 18;318(3):241–2.
- Eckel RH, Jakicic JM, Ard JD, de Jesus JM, Houston Miller N, Hubbard VS et al. 2013 AHA/ACC Guideline on lifestyle management to reduce cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014 Jun 24;129(25 Suppl 2):S76–99.
- Gregg EW, Williams DE, Geiss L.. Changes in diabetes-related complications in the United States, 1990–2010. N Engl J Med. 2014 Apr 17; 370(16):1514–23.
- Hift RJ. The porphyrias. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1408-16.
- Hirsh BJ, Smilowitz NR, Rosenson RS, Fuster V, Sperling LS.. Utilization of and adherence to guidelinerecommended lipid-lowering therapy after acute coronary syndrome: opportunities for improvement. J Am Coll Cardiol. 2015 Jul 14;66(2):184–92.
- Hooper L, Martin N, Abdelhamid A, Davey Smith G.. Reduction in saturated fat intake for cardiovascular disease. Cochrane Database Syst Rev. 2015 Jun 10; (6):CD011737.
- Hussain K. Hypoglicemia and pancreatic islet cell disorders. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1548-55.
- International Hypoglycaemia Study Group. Minimizing hypoglycemia in diabetes. Diabetes Care. 2015 Aug;38(8):1583–91.
- Inzucchi SE, Bergenstal RM, Buse JB, Diamant M, Ferrannini E, Nauck M et al. Management of hyperglycemia in type 2 diabetes, 2015: a patient-centered approach: update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care. 2015 Jan;38(1):140–9.
- Jakobsen GS, Småstuen MC, Sandbu R, Nordstrand N, Hofsø D, Lindberg M et al. Association of bariatric surgery vs medical obesity treatment with long-term medical complications and obesity-related comorbidities. JAMA. 2018 Jan 16;319(3): 291–301.
- Jensen MD. Obesitiy. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1458-66.
- Kalauz M., Vucelić B. Hemokromatoza. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008. str. 1283-6.
- Kalauz M., Vucelić B. Wilsonova bolest. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008. str. 1280-2.
- Kaler SG., Schilsky ML. Wilson disease. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1416-8.
- Karalis DG. A review of clinical practice guidelines for the management of hypertriglyceridemia: a focus on high dose omega-3 fatty acids. Adv Ther. 2017 Feb;34(2):300–23.
- Kim CH, Park JH, Park TS, Baek HS. Autoimmune hypoglycemia in a type 2 diabetic patient with anti-insulin and insulin receptor antibodies. Diabetes Care. 2004 Jan;27(1):288–9.
- Koršić M. Pagetova bolest kostiju i druge koštane displazije. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008. str. 1336-43.
- Koršić M., Giljević Z. Osteoporoza. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008. str. 1330-6.
- Koršić M., Misjak M. Fiziologija i dijagostika koštanog sustava. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008. str. 1318-21.
- Krasnewich DM., Sidransky E. Lysosomal storage diseases. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1399-403.
- Ma C, Avenell A, Bolland M, Hudson J, Stewart F, Robertson C et al. Effects of weight loss interventions for adults who are obese on mortality, cardiovascular disease, and cancer: systematic review and meta-analysis. BMJ. 2017 Nov 14; 359:j4849.
- Manary MJ, Trehan I. Protein-energy malnutrition. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1434-7.
- Mingrone G, Panunzi S, De Gaetano A, Guidone C, Iaconelli A, Leccesi L et al. Bariatric surgery versus conventional medical therapy for type 2 diabetes. N Engl J Med. 2012 Apr 26; 366(17):1577–85.
- Misjak M., Kaštelan D. Rahitis i osteomalacija. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008. str. 1321-4.
- Nauck M. Incretin therapies: highlighting common features and differences in the modes of action of glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors. Diabetes Obes Metab. 2016 Mar;18(3):203–16.
- Page MM, Watts GF. PCSK9 in context: a contemporary review of an important biological target for the prevention and treatment of atherosclerotic cardiovascular disease. Diabetes Obes Metab. 2018 Feb;20(2):270–82.
- Papa B. Poremećaji metabolizma porfirina. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008. str. 1298-302.
- Pereira GF, Bulik CM, Weaver MA, Holland WC, Platts-Mills TF. Malnutrition among cognitively intact, noncritically ill older adults in the emergency department. Ann Emerg Med. 2015 Jan;65(1):85–91.
- Reiner Ž. Bolesti lizosoma. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008. str. 1302-10.
- Reiner Ž. Poremećaji metabolizma lipida. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008. str. 1286-93.
- Reiner Ž. Poremećaji metabolizma purina. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008. str. 1293-7.
- Robinson JG, Nedergaard BS, Rogers WJ, Fialkow J, Neutel JM, Ramstad D et al; LAPLACE-2 Investigators. Effect of evolocumab or ezetimibe added to moderate- or high-intensity statin therapy on LDL-C lowering in patients with hypercholesterolemia: the LAPLACE-2 randomized clinical trial. JAMA. 2014 May 14;311(18):1870–82.
- Savage MW, Dhatariya KK, Kilvert A, Rayman G, Rees JA, Courtney CH et al. Joint British Diabetes Societies guideline for the management of diabetic ketoacidosis. Diabet Med. 2011 May;28(5):508–15.
- Schiff M., Blom H. Homocystinuria nad hyperhomocysteinemia. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1403-7.
- Scott AR; Joint British Diabetes Societies (JBDS) for Inpatient Care; JBDS hyperosmolar hyperglycaemic guidelines group. Management of hyperosmolar hyperglycaemic state in adults with diabetes. Diabet Med. 2015 Jun;32(6):714–24.
- Selph S, Dana T, Blazina I, Bougatsos C, Patel H, Chou R. Screening for type 2 diabetes mellitus: a systematic review for the U.S. Preventive Services Task Force. Ann Intern Med. 2015 Jun 2;162(11):765–76.
- Semenkovich CF. Dosorders of lipid metabolism. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1389-97.
- Stone NJ et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014 Jul 1;63(25 Pt B):2889–934.
- The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin dependentmdiabetes mellitus. N Engl J Med. 1993 Sep 30; 329(14):977–86.
- Thompson AE. JAMA patient page. Hypoglycemia. JAMA. 2015 Mar 24–31;313(12):1284.
- Vue MH, Setter SM. Drug-induced glucose alterations part 1: druginduced hypoglycemia. Diabetes Spectrum. 2011 Aug; 24(3):171–7.
- Waanders F, Visser FW, Gans RO. Current concepts in the management of diabetic nephropathy. Neth J Med. 2013 Nov;71(9):448–58.
- Weber TJ. Osteoporisis. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1637-45.
- Weinstein DA. Glycogen storage diseases. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1397-9.
- Weinstein RS. Osteomalacia and rickets. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1645-9.
- Yusuf S, Bosch J, Dagenais G, Zhu J, Xavier D, Liu L et al; HOPE-3 Investigators. Cholesterol lowering in intermediate-risk persons without cardiovascular disease. N Engl J Med. 2016 May 26;374(21):2021–31.
- Zjačić-Rotkvić V. Poremećaj sinteze i mobilizacije glikogena. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008. str. 1310-2.