BOLESTI KRVI I KRVOTVORNIH ORGANA
Klinički pristup bolesniku s bolestima krvi i krvotvornih organa
Bolesti matične hematopoetske stanice
Eritrocitopoeza i kliničko-patološki pojam anemije
Hipoproliferacijske anemije
Anemije zbog poremećaja u sazrijevanju eritrocita
Nasljedne hemolitičke anemije
Stečene hemolitičke anemije
Poremećaji broja i funkcije trombocita
Benigne bolesti i poremećaji leukocita
Kronične mijeloproliferativne bolesti
Mijelodisplastični sindrom
Maligne hematološke bolesti
Leukemije
Akutne leukemije Kronične leukemije
Maligni limfomi
Hodgkinov limfom
Ne-Hodgkinovi limfomi
Plazmastanične neoplazme
Fiziologija koagulacijskog sustava
Sklonost krvarenju ili hemoragijska dijateza
Sklonost zgrušavanju krvi ili trombofilija
Splenomegalija, hipersplenizam i ostale bolesti slezene
Transplantacija krvotvornih matičnih stanica
Hitna stanja u hematologiji
Literatura
KLINIČKI PRISTUP BOLESNIKU S BOLESTIMA KRVI I KRVOTVORNIH ORGANA
Klinički pristup bolesnicima oboljelih od bolesti krvi i krvotvornih organa podrazumijeva prepoznavanje osnovnih simptoma i znakova kao i temeljnih dijagnostičkih postupaka karakterističnih za ovu skupinu bolesti.
OSNOVNI SIMPTOMI I ZNAKOVI
Karakteristični hematološki sindromi koji se susreću u kliničkoj praksi su anemijski sindrom, pletora, hemoragijska dijateza te limfadenopatija i splenomegalija.
Anemijski sindrom nastaje kao posljedica sniženih vrijednosti broja eritrocita i hemoglobina u krvi, što dovodi do smanjene sistemske oksigenacije tkiva i organa. Intenzitet simptoma i znakova koje se povezuje s tim stanjem ovisi o više čimbenika, kao što su jačina anemije, duljina njezina trajanja (kronične anemije praćene su razvojem kompenzacijskih mehanizama) i opće stanje bolesnika. Bolesnici s umjereno izraženom anemijom (hemoglobin > 90 g/L) obično su asimptomatski, ukoliko nemaju dodatne komorbiditete (npr. kronična srčana ili bubrežna bolest i sl.). Glavne kliničke manifestacije anemijskoga sindroma obuhvaćaju bljedilo kože i sluznica, osjećaj umora i pospanosti, glavobolju i vrtoglavicu, osjećaj nedostatka zraka i lupanja srca uz različito izraženu tahikardiju. Ako se radi o anemijskom sindromu kao posljedici hemolitičke anemije, bolesnici su ikterični. Anemija može dovesti i do pogoršanja bolesti prisutnih od ranije, na primjer koronarne, cerebrovaskularne, periferne arterijske bolesti i sl. Klinički pristup oboljelima od anemije i njihova dijagnostička obrada opisani su u odgovarajućim poglavljima u daljnjem tekstu.
Pletora je naziv za skupinu kliničkih manifestacija koje nastaju kao posljedica povećanoga broja krvnih stanica te pojačane prokrvljenosti tkiva i organa. Najčešći je uzrok pletore apsolutno povećanje broja eritrocita (eritrocitoza), bilo da se radi o primarnoj (policitemija rubra vera) ili sekundarnoj, kao odgovoru na različita hipoksemična stanja, zatim relativno povećanje broja eritrocita (dehidracija, hemokoncentracija), a može se raditi i o povećanju broja drugih krvnih stanica. Najčešće kliničke manifestacije koje prate pletoru su zagasita, tamna i cijanotična boja kože i sluznica (osobito na periferiji) uz glavobolju, vrtoglavicu, osjećaj prepunjenosti glave i vrata, svrbež te pojava kožnih ulceracija. Kod takvih je bolesnika znatno češća pojava trombotskih incidenata, obično imaju povišene vrijednosti krvnoga tlaka te splenomegaliju. Temelj obrade takvih bolesnika čini obrada eritrocitoze i eventualno trombocitoze. S ciljem simptomatskoga olakšanja povremeno se takvim bolesnicima mogu izvoditi venepunkcije uz odstranjenje 200 do 400 ml krvi uz istodobnu nadoknadu iste količine infuzijskim otopinama (npr. fiziološka otopina).
Hemoragijska dijateza naziv je kojim se opisuju kliničke manifestacije i sklonost bolesnika nastanku krvarenja, što se povezuje s manjkavosti sustava zgrušavanja i/ili broja i funkcije trombocita. Ovisno o težini bolesti u podlozi, krvarenje kod takvih bolesnika može nastati spontano ili uz minimalnu traumu, ili se može očitovati smanjenom sposobnošću spontanoga zaustavljanja krvi (npr. nakon vađenja zuba, porezotina i sl.). Krvarenja se kod takvih bolesnika često očituju na koži i sluznicama (tzv. suha krvarenja), u obliku hematoma, sufuzija, petehija i ekhimoza. Vidljiva, palpabilna krvarenja u koži nazivaju se purpura, a mogu se naći kod bolesnika s vaskulitisom. Također, takvi su bolesnici skloni i krvarenju u različite šuplje organe i organske sustave (tzv. vlažna krvarenja), što se klinički može manifestirati kao hematemeza, melena, rektoragija, hematurija, uretroragija, hemoptiza, epistaksa i sl. Kod svih bolesnika sa sumnjom na hemoragijsku dijatezu obvezno je odrediti broj trombocita i obaviti osnovni koagulogram.
Limfadenopatija je naziv za povećanje limfnih čvorova, što se definira njihovim promjerom većim od 1 cm. U pravilu u odraslih osoba limfni čvorovi nisu palpabilni (osim u području prepona gdje im normalna veličina može biti i do 2 cm) te se svako njihovo uvećanje označava kao limfadenopatija. Uzroci limfadenopatije su različiti: upalno-reaktivna limfadenopatija (bakterijske, virusne i granulomatozne upale), maligna limfadenopatija (primarni tumori ili sekundarizmi), imunološka limfadenopatija (serumska bolest, bolesti vezivnoga tkiva, sarkoidoza) te druga specifična stanja (tezaurizmoze). Generalizirana limfadenopatija (limfadenopatija koja zahvaća tri ili više anatomskih regija) implicira sustavnu infekciju ili limfom, dok lokalizirana limfadenopatija ukazuje na specifična regionalna patološka oboljenja. Limfonodusi mekše konzistencije ukazuju na benignu narav, a tvrdi i fiksirani na malignu. Bolesnicima iznad 40 godina, onima s povećanjem supraklavikularnih i skalenskih čvorova te onima s bilo kojim čvorom većim od 4 cm treba napraviti što hitniju ekscizijsku biopsiju, dok je kod mlađih pacijenata, osobito onih s mekanim čvorovima, opravdano promatranje u trajanju od 7 do 14 dana (emprijska antibiotska terapija nije potrebna). Ako se čvorovi smanjuju, daljnja obrada nije potrebna, a ako se dalje povećavaju - indicirana je biopsija. Uz citološku punkciju i/ili biopsiju limfnog čvora s imunofenotipizacijom i histološkim opisom potrebno je učiti i serološke pretrage na CMV, EMV, Toxoplasmu gondii, dijagnostiku tuberkuloze te sarkoidoze (ACE enzim).
Splenomegalija je naziv za patološko uvećanje slezene koje može biti praćeno i njezinom povećanom funkcijom (hipersplenizam). Često se susreće kod bolesnika s hematološkim oboljenjima, a detaljno je opisana kasnije, u dijelu o bolestima slezene.
OSNOVNI DIJAGNOSTIČKI POSTUPCI
Osnovni dijagnostički postupci u hematologiji obuhvaćaju standardne hematološke i koagulacijske laboratorijske pretrage, specifične biokemijske hematološke pretrage, citomorfološku i histomorfološku dijagnostiku, imunološku fenotipizaciju te citogenetske i molekularne dijagnostičke metode.
Hematološke laboratorijske pretrage. Osnovne hematološke pretrage uključuju određivanje kompletne i diferencijalne krvne slike. Kompletnom krvnom slikom određuju se broj leukocita, broj eritrocita, vrijednosti hemoglobina, hematokrita, eritrocitnih indeksa (MCV, MCH, MCHC), širina distribucije volumena eritrocita (RDW), broj retikulocita, broj trombocita te prosječna veličina trombocita (MPV). Diferencijalna krvna slika obuhvaća određivanje pojedinih vrsta leukocita (neutrofili, eozinofili, bazofili, limfociti, monociti). U većini se laboratorija oni određuju u postocima, a ne u apsolutnim brojevima. Apsolutni broj pojedinih vrsta leukocita dobiva se tako da se dobiveni postotak podijeli sa 100 te pomnoži s ukupnim brojem leukocita (u jedinici x 109/L). Detaljnije je značenje i tumačenje pojedinih elemenata hematoloških pretraga opisano u odgovarajućim dijelovima udžbenika.
Koagulacijske laboratorijske pretrage. Standardni koagulogram obuhvaća određivanje sljedećih parametara: trombinsko vrijeme (TV), protrombinsko vrijeme (PV), aktivirano parcijalno tromboplastinsko vrijeme (aPTV), vrijednosti fibrinogena, antitrombina III i D-dimera. U dodatne koagulacijske pretrage ubrajaju se određivanje aktivnosti pojedinih čimbenika koagulacije, plazminogena i inhibitora plazmina, probir na inhibitore čimbenika zgrušavanja, test miješanja s normalnom plazmom te ispitivanje agregacije trombocita. Značenje i tumačenje pojedinih elemenata osnovnog i proširenog koagulograma opisani su u poglavlju „Fiziologija koagulacijskog sustava“.
Specifične biokemijske hematološke pretrage podrazumijevaju specifične pretrage koje se ciljano izvode kod određenih bolesnika i određenih indikacija, kao što su test autohemolize kod dokazivanja hereditarne sferocitoze, dokazivanje Heinzovih tjelešaca koja se vide kod bolesnika s deficitom glukoza-6-fosfat dehidrogenaze, određivanje koncentracije ili aktivnosti određenih enzima u eritrocitima (heksokinaze, glutation-reduktaze, piruvat-kinaze, glukoza-6-fosfat dehidrogenaze), određivanje globinskih lanaca u dijagnostici talasemija, određivanje patoloških oblika hemoglobina (karboksihemoglobin, sulfehemoglobin, methemoglobin) te određivanje eritropoetina. U specifične se biokemijske hematološke pretrage ubraja i ferogram koji podrazumijeva određivanje vrijednosti serumskoga željeza, ukupne i nezasićene sposobnosti vezivanja željeza, feritina i transferina, kao i određivanje vitamina B12 i folne kiseline u dijagnostici megaloblastičnih anemija.
Citomorfološka i histomorfološka dijagnostika čine temelj dijagnostike tumorskih bolesti krvi i krvotvornih organa. Oni se odnose na uzorke dobivene punkcijom, odnosno biopsijom limfnih čvorova ili koštane srži, rjeđe slezene. Citomorfologija ukazuje na broj i izgled stanica u uzorku dobivenom punkcijom, dok histomorfološki nalaz daje širi uvid u sastav i izgled uzorka, tj. tkiva u njemu, koji je dobiven biopsijom (koštana srž, limfni čvor).
Imunološka fenotipizacija krvotvornih stanica (imunofenotipizacija) predstavlja laboratorijsku dijagnostičku metodu koja omogućava otkrivanje specifičnih biljega na površini ili unutar krvotvornih stanica koje pripadaju skupini leukocitnih diferencijacijskih antigena, a nazivaju se još i CD-biljezi (engl. clusters of differentiation). Ta metoda ima jednu od najbitnijih uloga u dijagnostici, diferencijaciji i praćenju bolesnika s neoplazmama krvotvornoga sustava jer se određivanjem specifičnih CD-biljega omogućava identifikacija stanične loze (porijekla) stanica malignoga klona, kao i stadij njihove diferencijacije i zrelosti. Imunofenotipizacija krvotvornih stanica može se raditi na temelju različitih uzoraka (periferna krv, koštana srž, limfni čvor). Glavne laboratorijske metode koje omogućavaju provođenje imunofenotipizacije su imunofluorescencija te protočna citometrija.
Citogenetske i molekularne dijagnostičke metode omogućavaju otkrivanje specifičnih kromosomskih anomalija te određenih genskih mutacija koje, ne samo da imaju svoje mjesto u dijagnostici, nego i u planiranju liječenja te prognoze neoplastičkih bolesti krvotvornog sustava. Citogenetske metode (metode pruganja kromosoma, fluorescentna in situ hibridizacija, FISH) omogućuju uvid u broj i morfologiju kromosoma, odnosno omogućuju otkrivanje specifičnih kromosomskih anomalija, dok molekularne metode (PCR, engl. polymerase chain reaction) omogućuju analizu nukleinskih kiselina, odnosno otkrivanje specifičnih mutacija te njihove genske ekspresije.
BOLESTI MATIČNE HEMATOPOETSKE STANICE
Matična hematopoetska stanica predstavlja jedinstvenu pluripotentnu stanicu od koje nastaju sve zrele krvne stanice procesom koji se naziva hematopoeza. Matične stanice nalaze se u hematopoetskim tkivima (koštana srž) i karakterizira ih trajna sposobnost dijeljenja, što i osigurava stalni nastanak novih krvnih stanica. Proliferacijom pluripotentne matične hematopoetske stanice nastaju multipotentna mijeloidna i limfoidna matična stanica, a one ulaze u daljnji proces diferencijacije kojime nastaju sve ostale krvne stanice: T i B-limfociti iz limfoidne matične stanice te eritrociti, trombociti, monociti i granulociti iz mijeloidne matične stanice (Slika 6.1.). Normalno stvaranje i sazrijevanje krvnih stanica ovisi i o uvjetima okoliša u kojemu nastaju, kao i o prisutnosti čimbenika rasta (čimbenici koji potiču kolonije – CSF, engl. colony stimulating factors). Najznačajniji čimbenici koji potiču kolonije jesu G-CSF (potiče stvaranje granulocita), GM-SCF (potiče stvaranje granulocita i makrofaga), M-SCF (potiče stvaranje makrofaga), interleukin-3 (potiče stvaranje svih mijeloidnih stanica) te eritropoetin (potiče stvaranje eritrocita). U shematskom prikazu ispod teksta prikazan je proces hematopoeze, odnosno nastanak zrelih krvnih stanica iz matične pluripotentne stanice.
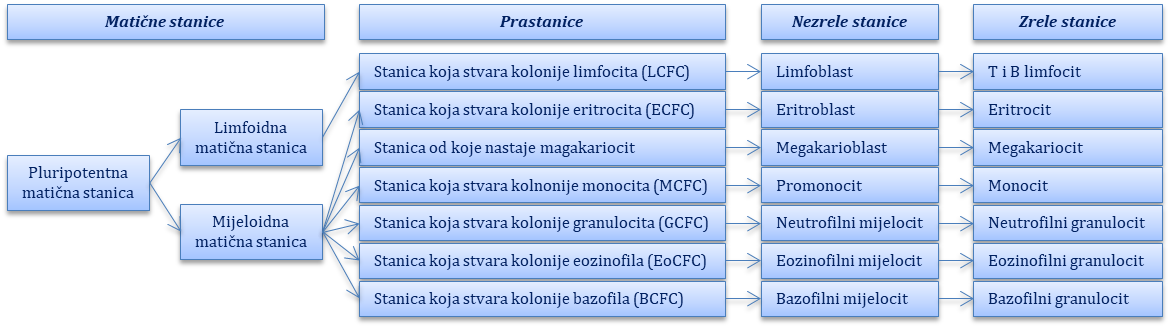
Slika 6.1. Shematski prikaz hematopoeze
Bolesti matične hematopoetske stanice. Glavne bolesti i poremećaji matične hematopoetske stanice su mijeloaplazije koje predstavljaju skupinu bolesti karakteriziranu potpunim ili djelomičnim manjkom pluripotentne matične hematopoetske stanice ili neke od usmjerenih matičnih stanica (limfoidna ili mijeloidna matična stanica), što za posljedicu ima smanjeno stvaranje odgovarajućih krvnih stanica. Glavne bolesti i poremećaji te skupine su aplastična anemija, izolirana aplazija crvene loze te paroksizmalna noćna hemoglobinurija. U bolesti matične hematopoetske stanice ubrajaju se i sindrom mijelodisplazije te akutne i kronične mijeloproliferativne bolesti, što je opisano zasebno.
APLASTIČNA ANEMIJA
Definicija. Aplastična anemija predstavlja primarni ili sekundarni poremećaj mijeloidne matične stanice koji rezultira njezinom smanjenom sposobnošću proliferacije, zbog čega dolazi do smanjenoga stvaranja stanica mijeloidne loze (pancitopenija) uz izraženu hipocelularnost koštane srži. U 20 % slučajeva radi se o nasljedno uvjetovanome poremećaju, dok se u 80 % slučajeva radi o stečenome stanju. Incidencija aplastične anemije u Europi iznosi oko 2 oboljela na milijun stanovnika, a bolest se podjednako javlja i kod muškaraca i žena. S obzirom na dobnu pojavnost, bolest pokazuje bifaznu raspodjelu s prvim vrškom pojavnosti u mlađoj životnoj dobi (između desete i dvadesete godine života) te drugim u starijoj životnoj dobi između šestoga i sedmog desetljeća života.
Etiopatogeneza. Aplastična anemija može se javiti u sklopu nasljednih poremećaja ili kao stečeno stanje. Najčešće nasljedne bolesti u sklopu kojih se razvija aplastična anemija su Fanconijeva anemija te kongenitalna diskeratoza. Uzroci stečene aplastične anemije najčešće ostaju nepoznati, pa govorimo o primarnoj ili idiopatskoj aplastičnoj anemiji, ili im uzrok može biti poznat, pa govorimo o sekundarnoj aplastičnoj anemiji.
Aplastična anemija u sklopu nasljednih poremećaja. Najčešći nasljedni poremećaji koji dovode do aplastične anemije su Faconijeva anemija i kongenitalna diskeratoza, a može se naći u sklopu drugih, rjeđih bolesti i stanja, kao što su obiteljska aplastična anemija te retikularna disgeneza. U takvim se slučajevima aplastična anemija najčešće razvija već u djetinjstvu.
Fanconijeva anemija je autosomno-recesivni poremećaj kod kojega je prisutna mutacija jednoga od FA gena (FA-A do FA-P) koje nalazimo na većem broju kromosoma, a njihovi genski produkti sudjeluju u procesima vezanim za staničnu diobu (kontrola stabilnosti molekule DNA, kontrola programirane stanične smrti). U više od 60 % slučajeva radi se o mutaciji FA-A gena koji se nalazi na dugom kraku 16. kromosoma. Klinički se bolest očituje uglavnom već do desete godine života, a glavne su kliničke karakteristike niži tjelesni rast, kožne hiperpigmentacije (pjege boje bijele kave, engl. cafe au lait spots), koštane abnormalnosti (osobito palca i palčane kosti) te genitourinarne anomalije (hipoplazija bubrega) uz određeni stupanj mentalne retardacije.
Kongenitalna diskeratoza predstavlja nasljedni poremećaj karakteriziran mutacijom gena čiji produkti sudjeluju u održavanju dužine telomera (krajnji dijelovi kromosoma), tako da one sa svakim dijeljenjem stanica postaju kraće te naposlijetku dovode do apoptoze. Najčešće se radi o mutaciji TERT gena (engl. telomerase reverse transcriptase gene). Kliničke karakteristike oboljelih uključuju leukoplakiju sluznica (najčešće oralna leukoplakija), distrofiju noktiju, hiperpigmentaciju kože (vrat i prsa) te razvoj aplastične anemije u ranom djetinjstvu.
Stečena aplastična anemija može imati nepoznat uzrok, pa se naziva primarna ili idiopatska aplastična anemija, ili može imati poznat uzrok, pa govorimo o sekundarnoj aplastičnoj anemiji.
Primarna (idiopatska) aplastična anemija To je ujedno i najčešći oblik aplastične anemije. Danas se smatra da je ona posljedica patološkoga imunološkog zbivanja, s obzirom na to da se u koštanoj srži oboljelih nalazi povećan broj aktiviranih citotoksičnih T-limfocita, kao i medijatora TH-1 odgovora (TNF-α, IFN-γ) za koje se pretpostavlja da uzrokuju apoptozu i destrukciju hematopoetske matične stanice. Točan uzrok aktiviranja takvoga imunološkog odgovora nije poznat, a smatra se da može biti potaknut nekim vanjskim ili vlastitim antigenom osoba koje su već genetski predisponirane.
Sekundarna aplastična anemija razvija se sekundarno, kao odgovor na određenu noksu ili u sklopu određene bolesti ili stanja. Najčešći uzroci sekundarne aplastične anemije su zračenje, lijekovi i toksini te infekcije.
Djelovanje X-zraka na hematopoetsku matičnu stanicu (npr. vanjsko zračenje) može dovesti do njezinoga oštećenja, što može rezultirati hipoplazijom ili aplazijom koštane srži, a težina ovisi o količini i duljini primjene zračenja, kao i individualnim razlikama u osjetljivosti na X-zrake.
Različiti kemijski agensi također mogu dovesti do oštećenja hematopoetske stanice. Takvo oštećenje može biti predvidivo i ovisno o dozi, ili nepredvidivo i neovisno o dozi (reakcija idiosinkrazije). Kemoterapeutici (antagonisti folne kiseline, alkilirajući lijekovi, antraciklini, analozi baza) obično dovode do predvidljive reakcije, a težina aplazije ovisi o dozi i duljini primjene lijeka. Karakteristično kod takvih bolesnika dolazi do oporavka koštane srži nakon izostanka primjene tih lijekova, iako ona u nekih bolesnika može i izostati. Najčešći lijek koji dovodi do aplazije koštane srži reakcijom idiosinkrazije je kloramfenikol, dok rjeđe to mogu biti i preparati sulfonilureje, soli zlata, fenitoin, karbamazepin te još neki. Derivati benzena i slični ugljikovodici također mogu dovesti do aplazije koštane srži, najprije do reverzibilne, no, ukoliko izlaganje potraje kroz dulje vrijeme, može se razviti i ireverzibilna aplazija koštane srži.
Aplastična anemija može se razviti nakon infektivnog hepatitisa što se obično događa kod mlađih osoba, i to unutar prva dva mjeseca od akutne faze bolesti. Pancitopenija kod njih može biti izrazito teška. Kao relativno česti uzročnici spominju se i Epstein-Barrov virus (nakon infekciozne mononukleoze) te parvovirus B19. I kod bolesnika s HIV infekcijom također se može naći određeni stupanj pancitopenije i hipoplazije koštane srži.
Klinička slika. U većini slučajeva bolest počinje postupno i neprimjetno, najprije znacima anemije i hemoragijske dijateze, a kasnije i učestalim infekcijama. Bolesnici su blijedi, žale se na slabost i malaksalost, imaju palpitacije, nedostatak zraka i zujanje u ušima. Težina anemijskih simptoma ovisi o težini bolesti i izražaju same anemije. Nekada su prvi znakovi bolesti znakovi hemoragijske dijateze, kao što su ekhimoze i petehije te krvarenja iz gingiva i epistakse. Moguća su i krvarenja iz probavnoga sustava, a kod žena obilne i produljene menstruacije (menometroragija). Rijetka su masivna i po život opasna krvarenja. Infekcija kao prvi znak kliničke manifestacije aplastične anemije izuzetna je rijetkost, unatoč izraženoj agranulocitozi. Ukoliko je infekcija prvi i dominantni znak pancitopenije, mala je vjerojatnost da je posrijedi aplastična anemija. U fizikalnom nalazu obično dominiraju znakovi anemije (bljedoća kože i sluznica) i znakovi hemoragijske dijateze (petehije, ekhimoze). Bolesnici obično nemaju hepatosplenomegaliju niti limfadenopatije. Kod sumnje na nasljedni oblik aplastične anemije treba obratiti pozornost na pojedina fenotipska obilježja, kao što su niži rast, hiperpigmetacije kože, malformacije skeleta i sl., ali je to manje vjerojatno u starijoj životnoj dobi.
Dijagnoza se postavlja na temelju laboratorijskih nalaza periferne krvi te nalaza punkcije i/ili biopsije koštane srži. U svrhu otkrivanja uzroka provode se i druge dijagnostičke metode. Na osnovi broja stanica u perifernoj krvi i nalaza biopsije koštane srži procjenjuje se težina aplastične anemije koju dijelimo u tri kategorije (Camitta kriteriji), kako je prikazano u Tablici 6.1.
U laboratorijskim nalazima periferne krvi nalazimo pancitopeniju različitoga stupnja. Rjeđe pri aplastičnoj anemiji nalazimo zahvaćenost jedne ili dvije od tri krvne loze, a ako se to i nađe, obično je to u početku bolesti. Anemija je praćena smanjenim brojem retikulocita, a eritrociti su uredne veličine ili mogu biti uvećani (povišen MCV). Neutrofili i trombociti su sniženi, ali se ne susreću nezrele ili abnormalne forme u razmazu periferne krvi. Broj limfocita je uredan.
Pregled koštane srži (biopsija) ukazuje na hipocelularnost sa smanjenim brojem hematopoetskih stanica (ispod 25 %) i povećanim brojem, tj. udjelom masnih stanica. U vrlo teškim oblicima bolesti gotovo da i nema hematopoetskih stanica, nego samo masna infiltracija. Preostale hematopoetske matične stanice u koštanoj srži uredne su morfologije.
Ostale pretrage korisne u procjeni uzroka ili diferenciranja pancitopenije su: citogenetske analize kod sumnje na nasljedne poremećaje, serološke pretrage kod dokazivanja virusne etiologije te protočna citometrija kod dokazivanja paroksizmalne noćne hemoglobinurije. Ultrazvuk ili CT abdomena koristan je u procjeni postojanja hepatosplenomegalije ili visceralne limfadenopatije. Scintigrafija koštane srži također može biti korisna pri procjeni smanjene funkcije krvotvornoga sustava.
|
Tablica 6.1. Procjena težine aplastične anemije
|
|
Umjerena aplastična anemija
|
Teška aplastična anemija
|
Izrazito teška aplastična anemija
|
|
Broj hematopoetskih stanica u uzorku koštane srži manji od 25 % (ili 25 – 50 % uz više od 30 % rezidualnih stanica) plus najmanje dva od sljedećih kriterija:
|
|
Neutrofili > 0,5 x 109/L
Trombociti > 20 x 109/L
Retikulociti > 20 x 109/L
|
Neutrofili < 0,5 x 109/L
Trombociti < 20 x 109/L
Retikulociti < 20 x 109/L
|
Neutrofili < 0,2 x 109/L
Trombociti < 20 x 109/L
Retikulociti < 20 x 109/L
|
Diferencijalna dijagnoza. Aplastičnu anemiju treba razlikovati od drugih uzroka pancitopenije. U Tablici 6.2. su navedeni mogući uzroci pancitopenije. Mijelodisplastični sindrom i akutna leukemija razlikuju se od aplastične anemije prema postojanju staničnih morfoloških abnormalnosti u koštanoj srži, kao i povećanjem broja blasta u perifernoj krvi. Leukemija vlastastih stanica prepoznaje se po prisutnosti splenomegalije, kao i abnormalnih limfoidnih stanica u bioptatu koštane srži. Pancitopeniju uz normocelularnu koštanu srž moguće je susresti kod sistemskog eritematoznog lupusa, diseminirane infekcije, hipersplenizma te nutritivnih poremećaja (manjak vitamina B12 i folne kiseline). Izolirana trombocitopenija može biti prvi znak aplastične anemije, što je moguće zamijeniti i tumačiti kao imunu trombocitopeniju.
|
Tablica 6.2. Etiologija pancitopenije
|
|
Poremećaji koji zahvaćaju koštanu srž
|
Poremećaji koji ne zahvaćaju koštanu srž
|
|
Aplastična anemija
Sindrom mijelodisplazije
Akutna leukemija
Kronična idiopatska mijelofibroza
Infiltrativne bolesti (limfom, mijelom, karcinom)
|
Splenomegalija i hipersplenizam
Sistemski eritematozni lupus
Infekcije (HIV, tuberkuloza, CMV, parvovirus B19)
Lijekovi i kemoterapeutici
Zračenje, nutritivni poremećaji
|
Liječenje. U liječenju aplastične anemije razlikuje se potporno te specifično liječenje bolesti. Sam terapijski pristup ovisi o težini bolesti, kao i o dobi bolesnika. Prvi i osnovni korak u liječenju je uklanjanje potencijalnoga uzroka aplastične anemije, ukoliko je on poznat.
Potporno liječenje bolesnika s aplastičnom anemijom podrazumijeva kontrolu simptoma i znakova uzrokovanih pancitopenijom. Anemija se ispravlja davanjem transfuzija eritrocita, a trombocitopenija primjenom transfuzija trombocita. Ipak, treba izbjegavati davanje transfuzija i svesti ih na najmanju mjeru kako bi se izbjegla senzibilizacija na odgovarajuće HLA antigene i tako smanjila mogućnost odbacivanja transplantata koštane srži ukoliko bi se išlo u liječenje tom metodom, a iz istoga se razloga ne preporuča davanje transfuzija krvnih pripravaka od užih članova obitelji (koji su potencijalni donori koštane srži). Preporuča se primjena koncentrata eritrocita siromašnih leukocitima, kao i transfuzija trombocita od istoga davatelja ukoliko je to moguće. Indikacija za primjenu transfuzije eritrocita je pad hemoglobina ispod 70 g/L, odnosno 90 g/L ukoliko se radi o kardiopatima. Današnje preporuke idu prema tome da se transfuzija eritrocita primjenjuje ukoliko su prisutni znaci anemične hipoksije, a ne kako bi se hemoglobin održavao unutar preporučenih vrijednosti. Transfuzije trombocita ordiniraju se kada trombociti padnu ispod 5 x 109/L. Kod sniženja broja granulocita i/ili pojave infekcija potrebna je specifična antimikrobna zaštita. Kod bolesnika s izraženom neutropenijom (< 0,5 x 109/L) provodi se sistemska profilaksa antibioticima širokoga spektra. U slučaju dokaza infekcije potrebno je odgovarajuće liječenje prema nalazu antibiograma.
Specifično liječenje podrazumijeva primjenu čimbenika koji stimuliraju stanice koštane srži, imunosupresivnih lijekova te transplantaciju koštane srži.
Primjena čimbenika koji stimuliraju stanice koštane srži. U ovu svrhu spominje se primjena androgena te specifičnih faktora rasta. Androgeni (oksimetolon, fluoksimetolon, nandrolon-dekanoat) popravljaju anemiju pojačavajući lučenje eritropoetina, a učinak se može očekivati u razdoblju od tri do šest mjeseci nakon početka uzimanja preparata. Primjenjivati se mogu i eritropoetski faktori rasta (epoetin, darbepoetin) te mijeloidni faktori rasta (filgrastim, sargramostim). Svi oni imaju manje izražen učinak i mogu se koristiti u blažim oblicima aplastične anemije.
S obzirom na to da je imunološki posredovano oštećenje hematopetske matične stanice najčešći i pretpostavljeni mehanizam stečene aplastične anemije, njegovo suprimiranje može dovesti do smanjenja toga učinka i omogućiti oporavak preostalih stanica u koštanoj srži. U tu se svrhu primjenjuje kombinacija antilimfocitnoga globulina (ALG) životinjskoga porijekla (konj, zec) i ciklosporina. Primjena ALG-a je parenteralna (spora intravenska infuzija) i traje pet do deset dana (15 - 40 mg/kg tijekom 6 - 8 sati). Preferira se primjena konjskog ALG-a u odnosu na zečji jer je primijećen bolji odgovor i bolje preživljenje. Primjenom životinjskoga seruma moguća je pojava infuzijske reakcije i serumske bolesti (febrilno stanje, kožne promjene, artalgije, trombocitopenija), a te se nuspojave preveniraju istodobnom primjenom metilprednizolona (0,5 - 1 mg/kg/dan). Primjenu kortikosteroida treba ograničiti na što niže doze i što kraće trajanje zbog moguće pojave avaskularne nekroze kostiju i zglobova. Ciklosporin se primjenjuje peroralno u punoj dozi (3 - 7 mg/kg/dan) tijekom šest mjeseci, a primjena prestaje ukoliko se postigne dobar odgovor. Početni odgovor na terapiju obično se očekuje nakon jednog do tri mjeseca liječenja, ali on nije potpun nego samo djelomičan. Zadovoljavajući odgovor očekuje se četiri do šest mjeseci od početka terapije i prisutan je u oko 60 % oboljelih. U slučaju relapsa bolesti može se ponovno primijeniti terapija ALG-om, ali u manjim dozama (3 - 5 mg/kg tijekom pet dana). Takav oblik liječenja primjenjuje se do tri puta (prva epizoda bolesti i dva relapsa bolesti). Istraživanje je utvrdilo da je dodavanje eltrombopaga (trombopoetinski agonist) standardnoj imunosupresivnoj terapiji povezano s boljim terapijskim odgovorom. Alogena transplantacija koštane srži učinkovita je mjera liječenja, osobito kod mlađih osoba. Primjenjuje se transplantacija od HLA kompatibilnoga donora, najčešće bliskog srodnika. Tim se postupkom postiže dugoročno preživljenje u djece i mlađih oboljelih u više od 90 % slučajeva. Isti postupak kod starijih polučuje lošije rezultate. Glavne su komplikacije odbacivanje transplantata i reakcija transplantata protiv primatelja (GvHD, engl. graft versus host disease).
Klinički pristup bolesniku s aplastičnom anemijom. Kod umjerene i blage aplastične anemije provode se potporne mjere uz eventualnu primjenu čimbenika koji stimuliraju stanice koštane srži (androgeni, mijeloidni i eritroidni faktori rasta). Kod teških oblika aplastične anemije kod mlađih osoba (ispod 30 godina) terapija izbora je alogena transplantacija koštane srži ukoliko postoji HLA kompatibilan donor, dok je kod starijih bolesnika prva linija liječenja imunosupresivna terapija, a u slučaju njezinog neuspjeha ide se na transplantaciju koštane srži.
Prognoza. Neliječena aplastična anemija završava smrću. Mlađe se osobe bolje oporavljaju, a kod malobrojnih može nastupiti i spontano izlječenje. Alogena transplantacija koštane srži HLA-kompatibilnoga donora poboljšava preživljenje za preko 80 % kod bolesnika ispod 20 godina i 65 – 70 % kod bolesnika između 20 i 50 godina. Primjena imunosupresivne terapije (ALG + ciklosporin) dovodi do povoljnoga učinka kod oko 70 % bolesnika. Kod jedne trećine bolesnika bilježi se relaps bolesti nakon imunosupresivne terapije. Kod četvrtine bolesnika se nakon više od deset godina praćenja pojavi druga klonalna hematološka bolest kao što je paroksizmalna noćna hemoglobinurija, akutna mijelična leukemija ili mijelodisplazija.
IZOLIRANA APLAZIJA CRVENE LOZE
Definicija. Izolirana aplazija crvene loze, za razliku od aplastične anemije kod koje nalazimo poremećaj mijeloidne matične stanice s posljedičnom pancitopenijom, predstavlja rijedak poremećaj usmjerene matične stanice za eritrocitopezu (stanica koja stvara kolonije eritrocita, ECFC) s posljedičnim nastankom normocitne i normokromne anemije uz održane vrijednosti ostalih krvnih stanica.
Etiopatogeneza. Slično kao i kod aplastične anemije, razlikuju se kongenitalni i stečeni oblik bolesti. Najčešći kongenitalni oblik izolirane aplazije crvene loze naziva se Diamond-Blackfanova anemija. Ona nastaje uslijed mutacije nekog od gena koji kodiraju proteine ribosoma (RPS7, RPS10, EPS17, RPS24, RPS26 i dr.), a udružena je i s drugim fenotipskim obilježjima (nizak rast, deformiteti trupa i ekstremiteta). Stečeni oblici izolirane aplazije crvene loze najčešće su povezani s timomom (5 – 15 %), infekcijom parvovirusom B19 koji pokazuje organotropiju prema razvojnim stanicama eritrocitne loze te imunološkim poremećajima (pretpostavljena supresija posredovana T-limfocitima). Izolirana aplazija crvene loze može biti povezana i s uzimanjem lijekova (fenitoin, trimetroprim-sulfametoksazol, mikofenolat mofetil, rekombinantni humani eritropoetin), autoimunim bolestima vezivnog tkiva te hematološkim malignim oboljenjima (kronična limfatična ili mijeloična leukemija, limfomi, mijelofibroza). Posljedično poremećaju usmjerene matične stanice za eritropoezu izostaje njezina normalna diferencijacija i sazrijevanje u parastanice te nezrele i zrele oblike eritrocita, zbog čega se razvija normocitna i normokromna anemija. S obzirom na to da su drugi oblici usmjerenih stanica uredni, ostaje očuvano stvaranje trombocita, monocita te granulocita.
Klinička slika ovisi o težini nastale anemije. U blažim oblicima bolesnici su asimptomatski, a anemija se otkrije slučajnim laboratorijskim nalazom. U težim oblicima bolesnici mogu razviti klasične znakove anemijske hipoksije (umor, malaksalost, palpitacije, dispneja) uz izraženu bljedoću kože i sluznica. Ovisno o etiologiji u kliničkoj slici, mogu se naći i kliničke manifestacije osnovne bolesti.
Dijagnoza se postavlja na temelju laboratorijskih nalaza te nalaza biopsije koštane srži. U laboratorijskim se nalazima pojavljuju normocitna i normokromna anemija te sniženi broj retikulocita uz uredan broj trombocita i leukocita. Biopsija koštane srži predstavlja glavnu dijagnostičku metodu za potvrđivanje izolirane aplazije crvene loze u kojoj nalazimo urednu granulocitopoezu, trombocitopoezu i limfopoezu uz nedostatak stanica crvene loze, odnosno nalaz tek pokoje nezrele stanice eritroidne loze. Diferencijalno-dijagnostički je potrebno isključiti anemiju kronične bolesti i druge oblike normocitnih anemija.
Liječenje se u prvom redu sastoji od izbjegavanja, suzbijanja ili liječenja osnovnog uzroka ukoliko je on poznat. Kod bolesnika s prisutnim znacima anemične hipoksije indicirana je primjena koncentrata eritrocita u svrhu simptomatskoga poboljšanja. Osnovu liječenja težih oblika čini primjena kortikosteroida u prvoj liniji liječenja (prednizon 60 mg/dan) uz dodatak ciklofosfamida (2 - 3 mg/dan), ukoliko se ne postigne odgovarajući odgovor na monoterapiju kortikosteroidima. U najtežim oblicima mogu se primijeniti i ciklosporin, humani imunoglobulini, azatioprin, antitimocitni globulin te rituksimab ili alemtuzumab.
PAROKSIZMALNA NOĆNA HEMOGLOBINURIJA
Definicija. Paroksizmalna noćna hemoglobinurija (PNH) predstavlja stečenu klonalnu bolest krvotvorne matične stanice koja nastaje uslijed mutacija pig-A gena s posljedičnom smanjenom ekspresijom glikozil-fosfatidil-inozitola (GPI) na membranama nastalih krvnih stanica zbog čega one postaju osjetljive na djelovanje komponenata komplementa s posljedičnom hemolizom. Opisanu smanjenu ekspresiju GPI-a nalazimo na svim stanicama (eritrociti, monociti, trombociti, granulociti), a očituje se perifernom pancitopenijom. U svojoj kasnijoj fazi paroksizmalna noćna hemoglobinurija prelazi u aplastičnu anemiju. Bolest je ime dobila prema prvim opažanjima povećane koncentracije hemoglobina u urinu kod ovih bolesnika tijekom noći (tamni urin), što je posljedica povećane aktivnosti komplementa i hemolize uslijed pojačane crijevne apsorpcije lipopolisaharida tijekom noći za koje se zna da djeluju kao snažni aktivatori komplementa.
Etiopatogeneza. Temeljni mehanizam nastanka bolesti označava mutacija pig-A gena smještenoga na kratkom kraku X-kromosoma, a odgovoran je za nastanak GPI-a iz N-acetil-glukozamina. Posljedično smanjenoj ekspresiji GPI-a na membranama krvnih stanica onemogućeno je vezanje drugih molekula za njihovu površinu među kojima se ističu CD55 (DAF, engl. deccay accelerating factor) te CD59 (MIRL, engl. membrane inhibitor of reactive lysis) koje imaju glavnu ulogu u inhibiranju učinka komponenata komplementa (C3b, C9) koji se vežu na površinu krvnih stanica. Posljedično manjku CD55 i CD59, na površini dolazi do pojačane destrukcije (citoliza) krvnih stanica zbog čega se razvija pancitopenija. Jačina hemolize ponajprije ovisi o stupnju smanjenja izražaja CD55 i CD59 na površini stanica. Osim hemolize, ovi su bolesnici skloni i stvaranju venskih tromboza što se povezuje s pojačanom aktivacijom trombocita uslijed djelovanja komplementa i posljedičnim pojačanim otpuštanjem prokoagulantnih tvari kao što su ADP i tkivni faktor, a trombozi su najsklonije splanhične vene (osobito hepatalne vene i portalna vena). Smanjenom protoku krvi kroz splanhične krvne žile pridonosi i činjenica da slobodni hemoglobin koji se oslobađa hemolizom ima sposobnost vezanja dušičnog oksida (NO) uslijed čega dolazi do povećane sklonosti vazokonstrikciji.
Klinička slika. Paroksizmalna noćna hemoglobinurija najčešće se očituje kao kronična hemolitička anemija praćena povremenim egzacerbacijama što je obično potaknuto stresnim stanjima (tjelesni napor, infekcije, kirurški zahvati, traume i sl.). Ovisno o izraženosti anemije bolesnici mogu imati karakteristične simptome (umor, malaksalost, palpitacije, nedostatak zraka, taman urin, ikterus). Venske tromboze povezane su s težim oblicima bolesti (bolesnici koji imaju veći postotak stanica sa smanjenim izražajem CD55 i CD59), a najčešće se očituje trombozom splanhičnih vena (hepatalne vene, vene porte, mezenterične vene), rjeđe dubokom venskom trombozom donjih ili gornjih ekstremiteta. Kod manjega broja bolesnika dolazi do prijelaza bolesti u aplastičnu anemiju, sindrom mijelodisplazije ili akutnu leukemiju.
Dijagnoza paroksizmalne noćne hemoglobinurije otežana je zbog rijetke pojavnosti i nespecifičnih kliničkih manifestacija. Na nju treba pomišljati kod svih bolesnika s nejasnom hemolitičkom anemijom i pancitopenijom, osobito u prisustvu tromboembolijskih incidenata. Osnovni laboratorijski nalazi prate obrazac hemolitičkih anemija (normocitna i normokromna anemija, retikulocitoza, hiperbilirubinemija, sniženi haptoglobin) uz različit stupanj leukopenije i trombocitopenije. Temelj dijagnoze predstavlja utvrđivanje postojanja PNH populacije stanica (tj. stanica sa smanjenom ili odsutnom ekspresijom CD55 i CD59) što omogućuje metoda protočne citometrije. Dodatno je primjenom fluorescentne boje aerolizina koja se veže za GPI molekule na membrani stanica moguće pratiti i njegov izražaj te sposobnost da na sebe veže proteine (FLAER metoda). U dijagnostičkom postupku od koristi može biti i Hamov test kojime se dokazuje pojačana osjetljivost eritrocita na komplement.
Liječenje ovisi o izražaju kliničke slike. Kod asimptomatskih bolesnika s umjereno izraženom hemolizom nije potrebno specifično liječenje. Specifično liječenje uključuje primjenu ekulizumaba, humanog monoklonalnog protutijela koje sprječava aktivaciju komplementa smanjujući tako hemolizu i trombozu. On se primjenjuje kod bolesnika koji imaju izraženu hemolizu (potrebe za učestalim transfuzijama), kao i kod bolesnika koji razviju tromboembolijski incident. Bez obzira na primjenu ekulizumaba, u akutnoj fazi trombotskog događaja primjenjuju se i ustaljene metode liječenja (antikoagulantno liječenje). Kod mladih je bolesnika moguća i alogena transplantacija koštane srži, a rezervirana je za bolesnike koji ne pokazuju odgovor na liječenje ekulizumabom.
ERITROCITOPOEZA I KLINIČKO-PATOLOŠKI POJAM ANEMIJE
Eritrocitopoeza je naziv za proces stvaranja i sazrijevanja eritrocita koji podrazumijeva staničnu diferencijaciju iz pluripotentne hematopoetske stanice, pri čemu eritrocit prolazi kroz različite razvojne faze u kojima se vrši sinteza hemoglobina, a eritrocit dobiva svoj konačni oblik. Ovisno o morfološkim karakteristikama tijekom eritrocitopoeze, nekoliko je razvojnih oblika eritrocita: proeritroblast, bazofilni eritroblast, polikromafilni eritroblast, ortokromafilni eritroblast, retikulocit te eritrocit. Tijekom toga procesa zbivaju se različite morfološke promjene koje uključuju smanjenje volumena stanice, nestajanje jezgre i jezgrice iz citoplazme (postupno smanjenje, zgušnjavanje kromatina te konačno izbacivanje iz citoplazme), smanjenje broja poliribosoma, nestajanje mitohondrija i drugih organela uz istodobno povećanje količine hemoglobina. Eritrocit u razvojnoj fazi retikulocita napušta koštanu srž te dospijeva u krvotok gdje se odvija konačno sazrijevanje u konačnu formu eritrocita (normalno, retikulociti čine oko 1 – 2 % ukupnoga broja eritrocita u perifernoj krvi).
Eritropoetin predstavlja glavni faktor rasta u procesu eritrocitopeze. Najvećim se dijelom stvara u bubrezima (90 %), dok se samo 10 % eritropoetina stvara u jetri. Nakon stvaranja i lučenja u krvotok njegovo vrijeme poluživota iznosi svega šest do devet sati, a njegovo glavno djelovanje odnosi se na CFU-E stanice (engl. colony forming unit – erythroid), vrstu hematopoetskih matičnih stanica, preteče proeritoblasta. Glavni poticaj za lučenje eritropoetina čini bubrežna hipoksija, odnosno smanjenje parcijalnoga tlaka kisika u bubregu što nalaže stvaranje novih eritrocita. Osim eritropetina koji ima glavnu ulogu u poticanju eritrocitopeze, ulogu mogu imati i neke druge molekule kao što su SCF (engl. stem cell factor), IL-3 (interleukin-3), GM-CSF (engl. granulocyte-macrophage colony stimulating factor), IGF-1 (engl. insulin like growth factor 1) i drugi.
Eritrociti (crvene krvne stanice) u svome konačnom obliku nemaju staničnu jezgru, citoplazma je gotovo u potpunosti ispunjena hemoglobinom, a cijeli eritrocit poprima bikonkavni izgled. Takav bikonkavni izgled osigurava veću površinu (povećani omjer površine u odnosu na volumen) potrebnu za bolju izmjenu i prijenos plinova (kisik, ugljični dioksid), što mu i je glavna zadaća u organizmu. Građa stanične membrane eritrocita u kojoj središnju ulogu imaju integralni membranski proteini (vezni protein 3, glikoforini, akvaporin) te periferni membranski proteini (spektrin, aktin, ankirin) osiguravaju priličnu savitljivost eritrocita, što ih čini pogodnima za prolazak kroz sitne kapilare. Unutrašnjost eritrocita ispunjena je hemoglobinom, glavnim prijenosnikom kisika i ugljičnog dioksida u krvi. Glavni izvor energije potrebne za funkcioniranje eritrocita je glukoza koja u njih može ući bez posredovanja inzulinom i anaerobnim se metabolizmom razgrađuje do laktata. U tom se procesu stvara i metabolit 2,3-BPG (bisfosfoglicerin) koji utječe na afinitet vezanja kisika za hemoglobin. Normalan životni vijek eritrocita u krvi traje oko 120 dana, a njegovo uklanjanje iz cirkulacije izravno je povezano s njegovim starenjem i promjenom morfoloških svojstava. Starenjem se savitljivost, odnosno fleksibilnost eritrocita postupno smanjuje pri čemu oni sve teže prolaze kroz kapilarnu mrežu, osobito u području slezene te pri tome pucaju i dolazi do njihova uništenja. Osim toga, staranjem dolazi i do gubitka enzima koji nadziru stvaranje energije iz glukoze, a molekula hemoglobina postupno denaturira. Sve to pridonosi zarobljavanju eritrocita u slezeni (u manjoj mjeri i u koštanoj srži) gdje oni bivaju sekvestrirani i fagocitirani od strane stanica retikuloendotelnoga sustava. Svaki dan se u prosjeku obnovi 1 % ukupne eritrocitne populacije.
Hemoglobin je ključni protein koji omogućava prijenos kisika i ugljičnog dioksida, a ispunjava unutrašnjost eritrocita. Hemoglobin se sastoji od hema i globina. Hem se sastoji od četiri protoporfirinska prstena koji u sredini sadrže željezo u fero-obliku koji na sebe može reverzibilno vezati kisik, dok globin predstavlja proteinski nosač hema koji se sastoji od četiri polipeptidna lanca od kojih su po dva u potpunosti jednaka. Kod odraslih dominira tzv. adultni hemoglobin koji se još naziva i hemoglobin A (HbA), a sastoji se od dva α i dva β lanca, dok se fetalni hemoglobin ili hemoglobin F (HbF) sastoji od dva α i dva γ lanca – on prevladava do šest mjeseci nakon rođenja kada se postupno zamjenjuje adultnim oblikom, te adultni hemoglobin A1 (HbA1) koji se sastoji od dva α i dva δ lanca prisutan u malom postotku (svega 2 – 3 % ukupne količine hemoglobina). U procesu sinteze jednu od ključnih, ali i limitirajućih uloga imaju željezo te vitamin B6, što je shematski prikazano na Slici 6.2. Vezanje i otpuštanje kisika s molekule hemoglobina uvjetovano je položajem polipeptidnih lanaca globina, tako da se ovisno o njihovim položajima razlikuju dva temeljna konformacijska oblika: T-oblik (tense, zategnuta konformacija) i R-oblik (relax, relaksirana konformacija). T-konformacija obilježena je manjim afinitetom hemoglobina prema kisiku, što omogućava njegovo lakše otpuštanje s hemoglobina, dok je R-konformacija obilježena većim afinitetom hemoglobina prema kisiku i dovodi do njegovoga težeg otpuštanja s hemoglobina. Otpuštanje kisika s hemoglobina (disocijacija) opisuje se disocijacijskom krivuljom, a ona ovisi, osim o već spomenutom 2,3-BPG-u, i o koncentraciji vodikovih iona (pH krvi) te tjelesnoj temperaturi. Povećana koncentracija 2,3-BDG-a, acidoza i povišena tjelesna temperatura pomiču krivulju disocijacije kisika udesno, što znači da smanjuju afinitet hemoglobina prema kisiku čime omogućuju njegovo lakše otpuštanje u tkiva. Hemoglobin oslobođen raspadom eritrocita metabolizira se u stanicama retikuloendotelnoga sustava, a dio ga se veže za haptoglobin u serumu (kada se potroši zaliha hapoglobina, u krvi se može naći i slobodni, nevezani hemoglobin). U tom procesu najprije dolazi do oksidacije hemoglobina u verdoglobin, iz kojega se oslobađaju željezo i globin, a ostatak hema mijenja svoj oblik u tetrapriolski lanac koji se naziva biliverdin. On se pod utjecajem biliverdin-reduktaze reducira u bilirubin koji se dalje metabolizira klasičnim putem.
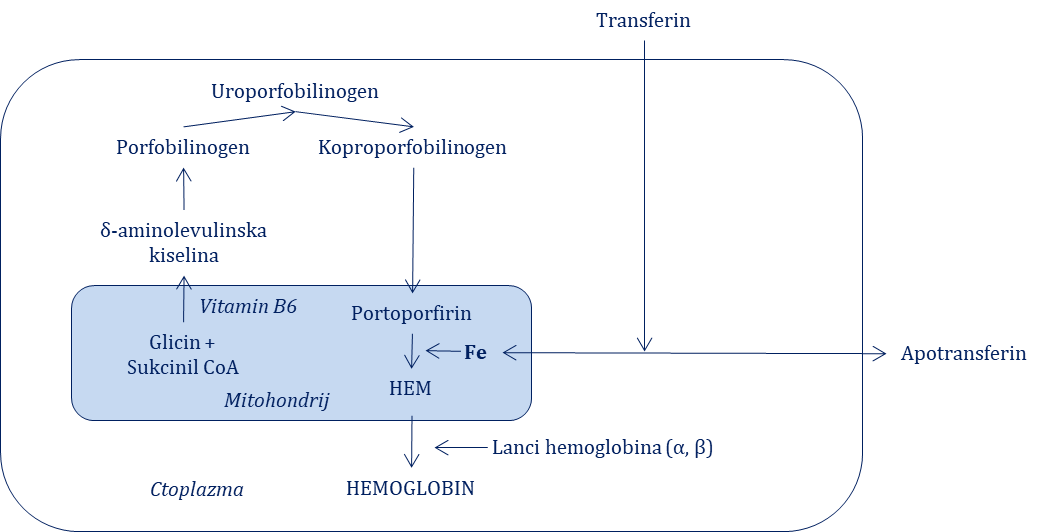
Slika 6.2. Shematski prikaz sinteze hemoglobina u eritroblastu.
Anemija ili slabokrvnost naziv je za stanje smanjenoga broja eritrocita, hemoglobina i/ili hematokrita u perifernoj krvi. Prema definiciji Svjetske zdravstvene organizacije definira se sniženim vrijednostima hemoglobina, i to u vrijednostima manjima od 130 g/L za muškarce i 120 g/L za žene. Kako referentne vrijednosti navedenih parametara ne ovise samo o ukupnoj količini eritrocita i hemoglobina nego i o njihovom odnosu prema volumenu plazme, anemija se može definirati i kao snižena sposobnost krvi za prijenos kisika (npr. u trudnoći se zbog povećanja volumena plazme smanjuje koncentracija eritrocita, hemoglobina i hematokrita uslijed dilucije, a ne stvarnoga smanjenja njihove mase, a obrnuto se događa u teško dehidriranih bolesnika). U kliničkoj se praksi obično koristi definicija zasnovana na vrijednostima hemoglobina uz poznavanje mogućih drugih utjecaja na njihove vrijednosti (npr. već spomenuta dehidracija, trudnoća i sl.).
Klasifikacija anemija može biti utemeljena na različitim osnovama, a najčešć je podjela anemija ovisno o kinetici stvaranja i nestajanja eritrocita te ovisno o morfologiji eritrocita.
Podjela anemija ovisno o kinetici podrazumijeva dvije skupine anemija: anemije koje nastaju uslijed smanjenoga stvaranja, povećane ili ubrzane razgradnje, ili uslijed gubitka eritrocita. Anemije uslijed smanjenoga stvaranja eritrocita mogu nastati ili zbog bolesti, odnosno poremećaja koštane srži (hipoproliferacijske anemije), ili uslijed poremećaja sazrijevanja eritrocita zbog manjkavosti tvari nužnih za njihov razvoj (željezo, vitamini B skupine). Anemije uslijed povećane ili ubrzane razgradnje nazivaju se hemolitičkim anemijama, mogu biti različite etiologije (intravaskularna ili ekstravaskularna hemoliza). Anemije uslijed gubitka eritrocita mogu se pojaviti u sklopu akutnoga ili kroničnog krvarenja. Akutno krvarenje rezultira brzim razvojem akutne hemoragijske anemije, dok kronično krvarenje rezultira sniženjem željeza u organizmu i posljedičnom anemijom uslijed poremećaja sazrijevanja eritrocita.
Podjela anemija ovisno o morfologiji zasniva se na veličini eritrocita koju se procjenjuje na temelju eritrocitnog indeksa MCV-a (engl. mean corpuscular volume) koji označava srednji volumen eritrocita. S obzirom na njegovu vrijednost, razlikuju se mikrocitne anemije (MCV < 80 fL), normocitne anemije (MCV 80 - 100 fL) te makrocitne anemije (MCV > 100 fL). Poznavanje te podjele ima značajno mjesto u kliničkoj praksi (diferenciranje uzroka ili mehanizma nastanka anemije).
U daljnjem razmatranju anemija i njezinih podvrsta, iz praktičnih će razloga one biti podijeljene u tri osnovne skupine utemeljene na mehanizmu nastanka anemije: hipoproliferacijske anemije, anemije zbog poremećaja u sazrijevanju eritrocita te hemolitičke anemije (nasljedne i stečene).
Kliničke manifestacije anemija. Na kliničku prezentaciju anemija ponajprije utječe brzina njezina razvoja, a potom i sama vrijednost hemoglobina. Spori razvoj anemije (kronična anemija) praćen je postupnim aktiviranjem kompenzatornih mehanizama (medularna i ekstramedularna hematopoeza, pomak disocijacijske krivulje hemoglobina udesno, povećanje srčanoga minutnog volumena), što umanjuje kliničku prezentaciju nastale anemije. Nagli razvoj anemije (akutna anemija) dovodi do jače izraženih kliničkih simptoma, upravo zbog manjkavosti kompenzatornih mehanizama. Kliničke manifestacije anemije mogu biti posljedica aktivacije kompenzatornih mehanizama (tahikardija, palpitacije, hepatosplenomegalija) ili sistemske tkivne hipoksije (umor, glavobolja, nestašica zraka, opresije u prsima, intermitentne klaudikacije i sl.). Dodatno, hemolitičke anemije mogu biti praćene razvojem ikterusa posljedično pojačanoj produkciji bilirubina koja nadilazi sposobnost (kapacitet) jetre da ga metabolizira. Prvi simptomi anemije obično se primijete tek kod pada vrijednosti hemoglobina ispod 90 g/L.
Laboratorijska dijagnostika anemija. Osnovna laboratorijska obrada bolesnika s anemijom uključuje određivanje crvene krvne slike, broja retikulocita te razmaz periferne krvi. Pojam crvena krvna slika podrazumijeva određivanje koncentracije eritrocita, hemoglobina, hematokrita, eritrocitnih indeksa (MCV, MCH, MCHC) te vrijednosti širine distribucije eritrocita. Ovisno o dobivenim nalazima i postavljanju sumnje na mogući uzrok anemije provode se dodatne laboratorijske i druge pretrage opisane u odgovarajućim dijelovima teksta.
Broj eritrocita i vrijednost hemoglobina temeljne su vrijednosti crvene krvne slike. Normalne vrijednosti eritrocita kreću se između 3,86 i 5,08x1012/L, dok se vrijednosti hemoglobina kreću između 120 i 160 g/L.
Hematokrit predstavlja postotak krvi koji čine eritrociti, a normalno iznosi od 41 do 53 % (ili 0,41 do 0,53). Kod tumačenja vrijednosti hematokrita treba imati na umu njegovu usku povezanost s volumenom plazme. Kod povećanja volumena plazme dolazi do razrjeđenja eritrocita, što dovodi do pada vrijednosti hematokrita, dok smanjenje volumena plazme dovodi do koncentriranja eritrocita, što dovodi do povišenja vrijednosti hematokrita.
Eritrocitni indeksi koji se standardno određuju su prosječni volumen eritrocita (MCV, engl. mean corpuscular volume), prosječni hemoglobin u eritrocitu (MCH, engl. mean corpuscular hemoglobin) te prosječna koncentracija hemoglobina u eritrocitu (MCHC, engl. mean corpuscular hemglobin concentration). Karakteristike i značenje eritrocitnih indeksa prikazani su u Tablici 6.3.
|
Tablica 6.3. Eritrocitni indeksi
|
|
Eritrocitni indeks
|
Normalne vrijednosti
|
Kliničko značenje
|
|
MCV
|
83,0 - 97,2 fL
|
Mjera obujma ili veličine prosječnog eritrocita
Omogućava razlikovanje mikrocitne, normocitne i makrocitne anemije
|
|
MCH
|
27,4 - 33,9 pg
|
Mjera za težinu hemoglobina u prosječnom eritrocitu
Omogućava razlikovanje normokromne i hipokromne anemije
|
|
MCHC
|
320 - 345 g/L
|
Mjera za koncentraciju hemoglobina u prosječnom eritrocitu
Omogućava iste informacije kao i MCH
|
Širina distribucije eritrocita (RDW, engl. red cell distribution width) označava mjeru za razliku između veličine eritrocita. U kliničkoj praksi bitno je uočavanje povećane vrijednosti širine distribucije eritrocita (> 15 %), što ukazuje na postojanje velike razlike između veličine cirkulirajućih eritrocita kakvo se može naći u određenim oblicima anemija.
Broj retikulocita ukazuje na sposobnost koštane srži da stvara nove eritrocite, a može se izraziti kao apsolutni broj ili kao postotak od ukupnoga broja eritrocita (klinički češće). Normalan udio retikulocita u odnosu na ukupni broj eritrocita u perifernoj krvi iznosi 1 do 2 %, a navedene se vrijednosti smatraju urednima kada se radi o bolesnicima koji nemaju anemiju. Kod bolesnika s anemijom kao kompenzacijski odgovor očekuje se pojačano stvaranje i otpuštanje retikulocita iz koštane srži u krv što mora dovesti do povećanja njihova udjela, a očekivani udio retikulocita kod anemičnih bolesnika iskazuje se formulom:
Sve vrijednosti udjela retikulocita ispod te izračunate vrijednosti smatraju se sniženima i ukazuju na neodgovarajući odgovor koštane srži, što se susreće u hipoproliferacijskim anemijama i anemijama zbog poremećaja sazrijevanja eritrocita. Nasuprot tome, povišene vrijednosti retikulocita (retikulocitoza) prisutne su kod bolesnika s hemolitičkim anemijama ili anemijama uslijed akutnog gubitka krvi (akutna hemoragijska anemija).
Razmaz periferne krvi omogućava uočavanje određenih morfoloških promjena eritrocita karakterističnih za određenu vrstu anemije. Ovisno o izgledu eritrocita promatranih pod svjetlosnim mikroskopom razlikuje se nekoliko njihovih morfoloških oblika, kao što su sferociti, mikrosferociti, ovalociti, dakrociti, kodociti, keratociti, shizociti, akantociti, drepanociti i dr. Opisi navedenih oblika i njihov dijagnostički značaj navedeni su u odgovarajućim dijelovima teksta.
HIPOPROLIFERACIJSKE ANEMIJE
Uvod. Hipoproliferacijske anemije nastaju uslijed bolesti ili poremećaja hematopoetske matične stanice, odnosno uslijed izostanka zadovoljavajuće diferencijacije stanica eritrocitne loze. U tu skupinu anemija ubrajaju se aplastična anemija, paroksizmalna noćna hemoglobinurija, izolirana aplazija crvene loze, anemija kronične bolesti, anemija kronične bubrežne bolesti, anemija endokrinoloških bolesti, anemija bolesti jetre te anemija uslijed infiltracije koštane srži. Aplastična anemija, paroksizmalna noćna hemoglobinurija i izolirana aplazija crvene loze opisane su u poglavlju „Bolesti matične hematopoetske stanice“.
ANEMIJA KRONIČNE BOLESTI
Definicija. Anemija kronične bolesti hipoproliferacijska je anemija koja se javlja kao posljedica kroničnih infektivnih i neinfektivnih bolesti (najčešće malignih) i zajedno sa sideropeničnom anemijom predstavlja najčešći oblik anemije u kliničkoj praksi. Anemija u sklopu kronične bolesti bubrega, anemija zbog endokrinoloških bolesti i bolesti jetre, kao i anemija uslijed infiltracije koštane srži izdvojene su iz ove skupine anemija zbog svoje različitosti u patogenetskom mehanizmu.
Etiopatogeneza. Anemija kronične bolesti najčešće se susreće u sklopu kroničnih infektivnih bolesti (tuberkuloza, plućni apsces, kronični pijelonefritis, osteomijelitis, subakutni bakterijski endokarditis), kroničnih autoimunih bolesti (sistemski eritematozni lupus, idiopatska crijevna upalna bolest, reumatoidni artritis i sl.) te malignih bolesti. Temeljni mehanizam nastanka te vrste anemije uključuje poremećeni metabolizam željeza povezan s povećanim stvaranjem proteina hepcidina. Hepcidin se pojačano stvara u hepatocitima kao odgovor na stimulaciju proupalnim citokinima, osobito interleukinom-6, djeluje na feroportin, transmembranski protein koji omogućuje transport željeza iz stanice u cirkulaciju. Vežući se za feroportin, dovodi do njegove inaktivacije i posljedičnoga zarobljavanja željeza u obliku feritina unutar stanice. To se ponajprije odnosi na makrofage koji sadrže veće količine željeza jer se u njima odvija razgradnja raspadnutih eritrocita i enterocita čime je onemogućena normalna apsorpcija željeza iz crijeva. Posljedično, željezo ostaje zarobljeno unutar stanica u obliku feritina (osobito u makrofazima i enterocitima), dok razina željeza u krvi ostaje smanjena. Osim toga, pri takvim bolestima neznatno je skraćen životni vijek eritrocita uslijed pojačane aktivnosti makrofaga, a prisutna je i relativna insuficijencija koštane srži uslijed inhibitornoga učinka proupalnih citokina, a sve to pridonosi razvoju ove vrste anemije.
Klinička slika i dijagnoza. Anemija kronične bolesti obično je blaga, s vrijednostima hemoglobina između 80 i 100 g/L, zbog čega rijetko daje klinička očitovanja i obično se dijagnosticira na temelju patološkoga laboratorijskog nalaza. Kliničkom slikom dominiraju simptomi i znaci osnovne bolesti. Anemija kronične bolesti razvija se kroz nekoliko mjeseci i nema tendenciju značajne progresije. Osnovni laboratorijski nalazi podrazumijevaju umjerenu mikrocitnu i normokromnu anemiju s nalazom ferograma koji pokazuje snižene vrijednosti serumskog željeza i transferina uz uredne ili povišene vrijednosti feritina. Pojašnjenje ferograma nalazi se u dijelu teksta o sideropeničnoj anemiji. U nejasnim slučajevima indicirana je koštana biopsija uz bojanje na željezo pri čemu se pojavljuje karakterističan nalaz izostanka bojanja željeza u sideroblastima (zbog nedostatka željeza) uz uredno ili pojačano bojanje željeza u koštanim makrofazima.
Diferencijalna dijagnoza. Glavna diferencijalno-dijagnostička dvojba kod ovih bolesnika uključuje razlikovanje anemije kronične bolesti od sideropenične anemije. Sideropenična anemija mikrocitna je i hipokromna, a u ferogramu uz snižene vrijednosti serumskog željeza nalazimo i snižene vrijednosti feritina i saturacije transferina, dok je vrijednost transferina povišena.
Liječenje. Osnovu liječenja anemije kronične bolesti čini liječenje bolesti u njezinoj podlozi. Transfuzije koncentriranih eritrocita rijetko su indicirane (kod vrijednosti hemoglobina nižih od 70 g/L), a nadomjesno liječenje željezom je kontraindicirano (osim u bolesnika kod kojih se primjenjuje liječenje eritropoetinom). Kod manjega broja bolesnika indicirana je i parenteralna primjena rekombinantnoga eritropetina (epoetin alfa, darbepoetin), i to prije svega kod kojih se ne može postići zadovoljavajući oporavak od anemije unatoč liječenju osnovne bolesti. Zajedno s primjenom eritropoetina indicirana je i parenteralna nadoknada željeza zbog pojačane potrebe za njim uslijed stimulacije eritrocitopeze (peroralna primjena nije svrsishodna zbog nemogućnosti odgovarajuće apsorpcije željeza uslijed djelovanja hepcidina). Ciljne vrijednosti hemoglobina kod takvih bolesnika su između 100 i 120 g/L jer je dokazana povezanost između primjene eritropoetina i povećanog rizika od nastanka tromboembolijskih incidenata, osobito kod bolesnika kojima hemoglobin poraste na više od 120 g/L.
ANEMIJA KRONIČNE BOLESTI BUBREGA
Definicija. Anemija kronične bolesti bubrega gotovo je redovita pojava, a nastaje poglavito zbog nedostatka eritropoetina uslijed bolesti bubrega. Na težinu anemije u prvom redu utječe stupanj oštećenja bubrežne funkcije, ali nije zabilježena povezanost između etiologije bubrežnoga oštećenja i težine anemije, osim u slučaju policistične bolesti bubrega (bolesnici s policističnom bolesti bubrega imaju nižu sklonost razvoju anemije).
Etiopatogeneza. Osnovni mehanizam nastanka anemije kod bolesnika s kroničnom bolesti bubrega smanjena je sposobnost bubrega da luči eritropoetin čija je osnovna zadaća stimulacija eritrocitopeze, stoga je ona oslabljena i nezadovoljavajuća. Drugi, dodatni čimbenici koji pospješuju razvoj anemije kod takvih bolesnika su: 1) inhibitorni učinak uremijskih toksina na funkciju koštane srži, 2) skraćeni životni vijek eritrocita uslijed metaboličkih i morfoloških promjena eritrocita izloženih uremijskim toksinima zbog čega su oni skloniji sekvestraciji u slezeni, 3) gubitak željeza i folne kiseline tijekom hemodijalize te 4) gubitak željeza uzrokovan kroničnim gubitkom krvi putem probavnoga i mokraćnog sustava (uremijski toksini inhibiraju aktivaciju trombocita što stvara predispoziciju za razvoj krvarenja). Posljedično navedenim mehanizmima stvaranje novih eritrocita je usporeno i nezadovoljavajuće, a stvoreni eritrociti kod većine su bolesnika normalnoga izgleda i volumena s urednom zastupljenosti hemoglobina (normocitna i normokromna anemija).
Klinička slika i dijagnoza. Anemija pri kroničnoj bolesti bubrega obično je asimptomatska i otkriva se na temelju patoloških laboratorijskih nalaza. U slučaju izražene anemije (vrijednosti hemoglobina ispod 70 g/L) mogu se pojaviti karakteristični znakovi anemije (umor, malaksalost, palpitacije, nedostatak zraka, glavobolje i sl.). U laboratorijskim nalazima očituju se normocitna i normokromna anemija što uz anamnestički podatak o kroničnoj bolesti bubrega ili patološkim nalazima dušikovih metabolita ukazuje na ovu vrstu anemije. Ovisno o prisutnosti dodatnih čimbenika, anemija može imati i obilježja megaloblastične ili sideropenične anemije. Broj retikulocita uredan je ili umjereno snižen, a vrijednosti serumskoga željeza, transferina i feritina variraju (pridružene kronične infekcije ili gubitak krvi pogoduju snižavanju serumskih vrijednosti željeza, dok učestale transfuzije mogu dovesti do preopterećenja željezom). Biopsija koštane srži rijetko se izvodi, a nalaz je gotovo uvijek uredan, što u kontekstu nalaza anemije predstavlja patološki nalaz jer izostaje nalaz eritroidne hiperplazije kao odgovora na anemiju.
Liječenje. Liječenje anemije pri kroničnoj bubrežnoj bolesti temelji se na primjeni rekombinantnih eritropoetina (epoetin, darbepoetin), a treba ga započeti kada vrijednosti hemoglobina padnu ispod 90 g/L. Početna doza za epoetin iznosi 50 jedinica/kg jednom ili dva puta tjedno, a za darbopoetin 0,45 mcg/kg svakih dva do četiri tjedna. Oba se primjenjuju subkutano ili intravenski s napomenom da je učinak za 30 % bolji nakon subkutane primjene. Glavna je nuspojava primjene rekombinantnih eritropoetina hipertenzija koja će zahtijevati ili smanjenje doze navedenih lijekova ili pojačanje antihipertenzivne terapije. Ostale, rjeđe komplikacije su encefalopatija i aplazija crvene loze, posredovane antieritropoetinskim antitijelima (češće kod primjene epoetina). Cilj je liječenja postići vrijednosti hemoglobina između 100 i 120 g/L (vrijednosti veće od 120 g/L nisu pokazale bolje kliničke ishode, a zabilježena je i veća pojavnost kardiovaskularnih komplikacija). Tijekom terapije se preporuča učestalije mjerenje krvnoga tlaka te praćenje vrijednosti hemoglobina svaka dva do četiri tjedna (očekivani porast hemoglobina kreće se oko 10 g/L kroz tri do četiri tjedna). Prije uvođenja ovih lijekova potrebno je odrediti ferogram i vitamine B skupine. U slučaju njihova deficita najprije se mora provesti supstitucija za njih (peroralno ili intravensko željezo, vitamini B skupine). U slučaju slaboga odgovora na navedenu terapiju treba razmišljati o drugim mogućim uzrocima anemije, kao i o nastanku antieritropoetinskih antitijela. Korekcija anemije kod takvih bolesnika dokazano poboljšava kvalitetu života, poboljšava toleranciju fizičkog napora te kognitivne funkcije.
ANEMIJA ENDOKRINOLOŠKIH BOLESTI
Uvod. S obzirom na dokazani učinak određenih hormona kao što su tiroksin, glukokortikoidi, testosteron i hormon rasta na diferencijaciju i sazrijevanje stanica eritroidne loze u koštanoj srži, za očekivati je da njihov deficit uzrokovan endokrinološkim bolestima može rezultirati nastankom određenoga stupnja anemije. Anemije endokrinoloških bolestih obično su umjerenoga izražaja, a nastali eritrociti normalnoga su volumena i sadržaja hemoglobina (normocitna normokromna anemija).
Hipotireoidizam. Kod bolesnika s manjkom hormona tiroksina može nastati umjerena normocitna anemija kao posljedica suprimirane eritrocitopeze, ali moguća je i pojava megaloblastične anemije i sideropenične anemije. Megloblastična anemija nastaje uslijed deficita vitamina B povezanog s pernicioznom anemijom koja je često prisutna uz hipotireozu, dok se sideropenična anemija može javiti kod žena s izraženim menoragijama koje čine dio kliničke slike hipotireoze. Poremećaj rada štitne žlijezde treba uzeti u obzir kod svih bolesnika s nerazjašnjenom etiologijom anemije.
Insuficijencija adrenokorteksa. Anemija kod bolesnika s insuficijencijom kore nadbubrežne žlijezde nastaje uslijed deficita glukokortikoida i time suprimirane eritrocitopoeze što rezultira umjerenom normocitnom i normokromnom anemijom. Nastala anemija često je prikrivena zbog istodobne deplecije volumena plazme te se anemija otkriva tek nakon uvođenja supstitucijske terapije kada dođe do reekspanzije volumena plazme.
Hipogonodizam. Manjak androgena, poglavito testosterona, može rezultirati blagom hipoproliferacijskom, normocitnom i normokromnom anemijom. Učinak androgena na eritrocitopezu očituje se već fiziološkom razlikom u vrijednostima hemoglobina između muškaraca i žena.
Hipopituitarizam. Razvoj anemije kod bolesnika s hipopituitarizmom uvjetovan je smanjenjem stvaranja i izlučivanja prethodno navedenih hormona (tiroksin, glukokortikoidi, androgeni), zbog deficita hormona prednjeg režnja hipofize koji upravljaju radom ostalih žlijezda. Kao i ostale endokrine anemije, i ova anemija je blaga, normocitna i normokromna.
Klinički pristup. S obzirom na to da su anemije endokrinoloških bolesti blage ili umjerene, rijetko imaju kliničke manifestacije te ih se dijagnosticira na temelju patoloških laboratorijskih nalaza. Liječenje uključuje odgovarajuću hormonsku supstituciju nakon čega se korigira i sama anemija.
ANEMIJA BOLESTI JETRE
Kronična bolest jetre gotovo je uvijek povezana s blagom do umjerenom normocitnom anemijom, rjeđe makrocitnom, uz pojavu karakterističnih oblika eritrocita u perifernom razmazu krvi (engl. target cell, stomatociti). Točan mehanizam nastanka anemije nije poznat, ali poznato je da se nakon uspostave ponovne funkcije jetre (oporavak, transplantacija) oporavlja i anemija. Alkoholna bolest jetre dovodi do razvoja megaloblastične (makrocitne) anemije što je povezano s malapsorpcijom vitamina B skupine, kao i s izravnim inhibitornim učinkom alkohola na koštanu srž. Rjeđe kod takvih bolesnika može prisutna biti i sideropenična anemija kao posljedica kroničnog gubitka krvi putem probavnoga sustava.
ANEMIJA ZBOG INFILTRACIJE KOŠTANE SRŽI (MIJELOFTIZIČNA ANEMIJA)
Definicija. Anemija zbog infiltracije koštane srži ili mijeloftizična anemija nastaje uslijed infiltracije koštane srži drugim tkivima, najčešće tumorskim ili upalnim supstratom. Posljedično dolazi do narušavanje normalne građe i funkcije koštane srži što se osim anemijom može očitovati i nezadovoljavajućom produkcijom drugih krvnih stanica (pancitopenija).
Etiopatogeneza. Tri su najčešća uzroka nastanka mijeloftizične anemije: 1) infiltracija koštane srži tumorskim tkivom (metastatski karcinom dojke, prostate, pluća, štitnjače, želuca i kolona te melanoma i neuroblastoma), 2) infiltracija koštane srži granulomatoznim tkivom (tuberkuloza, sarkoidoza) te 3) infiltracija koštane srži u sklopu metaboličkih bolesti nakupljanja (glikogenoze, sfingolipidoze, mukopolisaharidoze). Anemija kod ovih bolesnika nastaje uslijed smanjenja mase hematopoetskoga tkiva koštane srži uslijed infiltracije stranim tkivom, kao i zbog same endokrinološke parakrine aktivnosti koja podrazumijeva lučenje različitih medijatora koji dovode do supresije eritrocitopeze u preostalom hematopoetskom tkivu, a i onih koji potiču razvoj fibroze koštane srži. Zbog anatomski narušene barijere između krvnih žila i koštane srži dolazi do otpuštanja nezrelih staničnih oblika u krvotok što se očituje leukoeritroblastozom.
Klinička slika i dijagnoza. Kliničke manifestacije anemije u prvom redu ovise o stupnju njezinoga izražaja, ali i o samoj uzročnoj (osnovnoj) bolesti koja je dovela do njezinoga nastanka. Dijagnoza mijeloftizične anemije postavlja se na osnovi laboratorijskih nalaza, slikovnih koštanih metoda te biopsijom koštane srži. U laboratorijskim nalazima obično je prisutna različito izražena normocitna i normokromna anemija uz pojavu različitih morfoloških oblika eritrocita u razmazu periferne krvi (dakriociti, poikilociti). U diferencijalnoj krvnoj slici različito su zastupljeni nezreli oblici stanica eritrocitne i granulocitne loze (leukoeritroblastoza). Radiološka i nuklearno-medicinska (scintigrafija) koštana dijagnostika može upućivati na patološku pregradnju u kostima (osteolitička i osteoplastična žarišta) što može neizravno ukazivati na ovaj oblik anemije. Biopsija koštane srži ključna je dijagnostička metoda za dokazivanje mijeloftizične anemije koja omogućava izravan uvid u infiltraciju koštane srži stranim tkivom (zbog lokaliziranoga procesa preporuča se uzimanje više uzoraka materijala s različitih lokalizacija). Citološka punkcija koštane srži nema veći značaj za dijagnostiku ovoga poremećaja.
Liječenje se sastoji od liječenja uzročne bolesti, a uz nju je povezana i sama prognoza. Simptomatsko liječenje podrazumijeva povremene transfuzije koncentriranih eritrocita. Primjena eritropoetina rijetko je indicirana.
ANEMIJE ZBOG POREMEĆAJA U SAZRIJEVANJU ERITROCITA
Uvod. Anemije zbog poremećaja u sazrijevanju eritrocita među najčešćim su vrstama anemija koje se susreću u kliničkoj praksi, a nastaju uslijed manjka željeza (sideropenična anemija) ili uslijed manjka ili poremećaja metabolizma vitamina B skupine (megaloblastična anemija).
SIDEROPENIČNA ANEMIJA (ANEMIJA ZBOG MANJKA ŽELJEZA)
Definicija. Sideropenična anemija predstavlja anemiju nastalu zbog poremećaja u sazrijevanju eritrocita uslijed nedostatka željeza, pri čemu nastaju eritrociti malog volumena i niskoga sadržaja hemoglobina (normocitna, hipokromna anemija). Radi se o najčešćem obliku anemije s procijenjenom prevalencijom od 1 do 2 % u odrasloj općoj populaciji.
Etiopatogeneza. Uzroci smanjene količine željeza (sideropenija) u organizmu mogu biti različiti, a dijele se u četiri temeljne skupine: manjak željeza u prehrani, poremećaj apsorpcije željeza, povećana potreba za željezom te kronični gubitak krvi (Tablica 6.4.). Za bolje razumijevanje etiopatogeneze sideropenične anemije potrebno je poznavati osnove metabolizma željeza što je opisano u poglavlju „Poremećaji metabolizma željeza“.
|
Tablica 6.4. Etiologija sideropenije
|
|
Manjak željeza u prehrani
|
Poremećaj apsorpcije željeza
|
Povećana potreba za željezom
|
Kronični gubitak krvi
|
|
Mliječna preharana;
Jednolična dugotrajna ishrana siromašna željezom.
|
Atrofični gastritis;
Sindrom malapsorpcije;
Stanje nakon resekcije želuca i/ili dvanaesnika.
|
Djetinjstvo;
Adolescencija;
Trudnoća;
Laktacija.
|
Probavni sustav (ulkus, tumori, angiodisplazije);
Maternica (menoragija);
Ostali uzroci (hemoptize, hematurija).
|
Najčešći uzrok sideropenije u odrasloj populaciji kod muškaraca su krvarenja iz probavnoga sustava. Zbog toga je pojava sideropenične anemije kod odraslih muškaraca visoko suspektna na malignu bolest probavnoga sustava, dok je kod žena najčešći uzrok kronični gubitak krvi putem obilnih menstruacija (menoragija).
Razvoj sideropenije i sideropenične anemije postupan je i polagan. Početno sniženje serumskih vrijednosti željeza nadoknađuje se povlačenjem željeza iz zaliha u stanicama retikuloendotelnoga sustava, enterocita i hepatocita u kojima se željezo nalazi uskladišteno u obliku feritina i hemosiderina. Taj se period naziva latentnom sideropenijom, a laboratorijski je obilježen urednim vrijednostima željeza u serumu uz snižene vrijednosti feritina. S vremenom se troše postojane zalihe željeza iz navedenih stanica, što rezultira sniženjem vrijednosti željeza u serumu, odnosno razvojem manifestne sideropenije.
Posljedično manjku željeza dolazi do poremećaja sinteze hema i sazrijevanja eritrocita zbog čega nastaje sideropenična anemija karakterizirana eritrocitima malog volumena sa sniženom koncentracijom hemoglobina u citoplazmi (mikrocitna i hipokromna anemija). Umjesto željeza, u procesu sinteze hema na njegovo mjesto može se vezati cink pri čemu nastaje kompleks cink-portoporfirin..
Klinička slika. Kliničke manifestacije anemije ovise o njezinoj težini, odnosno vrijednostima hemoglobina. Često su one klinički asimptomatske te se dijagnosticiraju kao slučajni patološki laboratorijski nalaz. Bolesnici s izraženom anemijom mogu se tužiti na slabost, malaksalost, osjećaj zaduhe pri naporima, palpitacije i glavobolje. Pri fizikalnom su pregledu dominantni znakovi smanjena prokrvljenost kože i sluznica (bljedoća) te umjerena tahikardija u mirovanju. Sideropenija može biti povezana i s nastankom poremećaja epitelnih stanica što se može manifestirati kao angularni stomatitis, koilonihija (krhki i deformirani nokti) te disfagija i hipoklorhidrija kao rezultat afekcije jednjaka i želuca. Trijas simptoma koji čine disfagija, glositis i hipokromna anemija naziva se Plummer-Vinsonov sindrom, a nastaje kao posljedica sideropenije.
Dijagnoza sideropenične anemije postavlja se na osnovi patoloških laboratorijskih nalaza, dok diferenciranje točnog uzroka zahtijeva temeljitu anamnezu i širu dijagnostičku obradu.
Klasični laboratorijski nalazi koji ukazuju na sideropeničenu anemiju su: 1) mikrocitna i hipokromna anemija (snižene vrijednosti eritrocita i hemoglobina uz snižene vrijednosti MCV-a i MCH-a); 2) snižene vrijednosti serumskoga željeza; 3) povišene vrijednosti transferina (TIBC) i njegovoga nezasićenog dijela (UIBC) uz smanjenu saturaciju transferina željezom (< 10 %) te 4) snižene vrijednosti feritina. Pri tumačenju vrijednosti feritina treba imati na umu da se njegove vrijednosti povisuju i do tri puta u stanjima kroničnih upalnih procesa te se kod njih stvarna vrijednost feritina dobiva tako da se izmjerena vrijednost feritina podijeli sa tri. U novije se vrijeme manjak željeza može iskazati i mjerenjem vrijednosti transferinskih receptora u serumu (sTfR) čija se vrijednost povećava u bolesnika sa sideropenijom te mjerenjem koncentracije hemoglobina u retikulocitima (vrijednosti manje od 26 pg/stanici visoko su suspektne na sideropeniju). Referentne vrijednosti i pojašnjenja osnovnih parametara ferograma prikazani su u Tablici 6.5. Odnos vrijednosti serumskoga željeza, transferina, saturacije transferina te TIBC i UIBC predstavljen je shematski.
|
Tablica 6.5. Tumačenje nalaza ferograma
|
|
Parametar
|
Referentna vrijednost
|
Značenje
|
|
Serumsko željezo
|
8,0 - 30,0 umol/L
|
Vrijednost željeza u serumu, podložna dnevnim oscilacijama
|
|
Transferin
|
2,00 - 3,60 g/L
|
Ukupna vrijednost transferina u serumu (prijenosnik željeza)
|
|
TIBC*
|
36,0 - 75,0 umol/L
|
Ukupni kapacitet vezanja željeza za transferin
|
|
UIBC**
|
26,0 - 59,0 umol/L
|
Nezasićeni kapacitet vezanja željeza za transferin
|
|
Saturacija transferina
|
16 – 45 %
|
Postotak željeza vezan za transferin i dostupan eritrocitopezi
|
|
Feritin
|
10,0 - 120,0 ug/L
|
Skladišni oblik željeza, procjena rezerve željeza u organizmu
|
*engl., Total Iron Binding Capacity, ** engl., Unsaturated Iron Binding Capacity
Odnos vrijednosti serumskog željeza, transferina, saturacije transferina te TIBC (Total Iron Binding Capacity) i UIBC (Unsaturated Iron Binding Capacity ) dana je shematski na Slici 6.3.
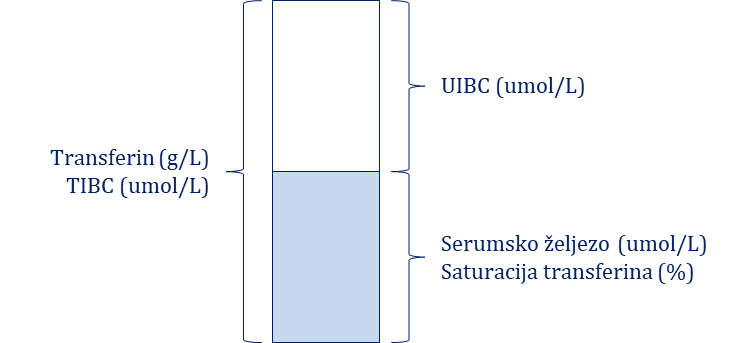
Slika 6.3. Shematski prikaz odnosa parametara ferograma
Daljnja dijagnostička obrada uzroka sideropenije i sideropenične anemije temelji se na anamnestičkim podacima i pridruženoj kliničkoj slici. Kod svih je bolesnika temeljni zadatak pri obradi sideropenične anemije isključiti okultno krvarenje u probavnom sustavu što obično nalaže izvođenje testiranja stolice na okultno krvarenje te endoskopsku obradu probavnoga trakta (gastroskopija, kolonoskopija).
Diferencijalna dijagnoza. Osim sideropenične anemije, mikrocitna hipokromna anemija može se naći u sklopu anemije kronične bolesti, talasemija te sideroblastične anemije. Glavana razlika između anemije kronične bolesti i sideropenične anemije leži u vrijednosti feritina koji je uredan ili povišen u anemiji kronične bolesti, dok je snižen u sideropeničnoj anemiji. Kod bolesnika s talasemijom vrijednosti ferograma su uredne, a u razmazu periferne krvi nalaze se tipični oblici eritrocita u obliku mete (engl. target cell). Sideroblastičnu anemiju karakteriziraju uredne ili povišene vrijednosti serumskog željeza, saturacije transferinom i feritina, uz tipičan nalaz prstenastih sideroblasta u uzroku koštane srži.
Liječenje. Osnovu liječenja sideropenične anemije čini liječenje osnovne bolesti odnosno njezinog uzroka. Korekcija sideropenične anemije provodi se peroralnom ili parenteralnom nadoknadom željeza.
Peroralna primjena željeza osnovni je način nadoknade željeza kod sideropeničnih bolesnika, ukoliko je očuvana apsorpcijska funkcija želuca i tankoga crijeva. U tu se svrhu najčešće primjenjuju fero-sulfat (3 x 325 mg/dan), fero-glukonat (2 – 3 x 300 mg/dan) te fero-fumarat (2 – 3 x 325 mg/dan). Apsorpcija je bolja ukoliko se preprati uzimaju na prazan želudac, no to je često praćeno većom učestalošću nuspojava (mučnina, dispepsija, opstipacija, dijareja, crne stolice), zbog čega se češće uzimaju zajedno s obrokom, ali je onda i apsorpcija slabija. Apsorpciju željeza iz probavnoga sustava smanjuju tein, kofein, antacidi, tetraciklini, oralni kontraceptivi, cink i infekcija H. pylori. Vitamin C pomaže apsorpciju željeza iz probavnog sustava, ali povećava i učestalost nuspojava, pa se ne koristi rutinski. Početak oporavka anemije može se očekivati kroz dva do tri tjedna od početka primjene peroralnih preparata željeza, a za potpunu normalizaciju nalaza potrebno je i do dva mjeseca terapije. Nakon normalizacije laboratorijskih nalaza terapija željezom trebala bi se provoditi kroz još tri do šest mjeseci (obnova zaliha željeza).
Parenteralna primjena željeza primjenjuje se kod bolesnika s razvijenim malapsorpcijskim sindromom, bolesnika koji ne podnose peroralnu primjenu željeza (izražene probavne smetnje) te kod bolesnika kod kojih se ne postiže ispravak sideropenije uz peroralnu nadoknadu željeza. Tijekom povijesti su se pripravci primjenjivali s posebnim oprezom zbog reakcija preosjetljivosti (uključujući anafilaksiju) koje bi nastajale pojačanim otpuštanjem željeza na mjestu primjene lijeka. Prijašnji intravenski pripravci željeza s monosaharidnim ili disaharidnim nosačem (željezov glukonat ili željezova sukroza) primjenjuju se nakon testne doze u maksimalnoj dozi od 125 do 200 mg/dan. Ti se pripravci mogu primjenjivati svakodnevno, a potrebno je više primjena. Novi pripravci željeza (željezova karboksimaltoza i željezov izomaltozid) primjenjuju se bez testne doze jednokratno ili u dvije primjene, što smanjuje broj dana provedenih na odjelu ili u dnevnoj bolnici. Učinkovitost se postiže zahvaljujući stabilnom ugljikohidratnom nosaču, kontroliranom otpuštanju željeza i njegovim visokim iskorištavanjem. Izračun potrebne doze intravenskoga željeza vrši se prema formuli: doza Fe (mg) = tjelesna težina (kg) x (ciljni hemoglobin – aktualni hemoglobin) x 0,22 + 1000 (za muškarce) ili 500 (za žene). Međutim, postoji pojedostavnjena shema prema vrijednostima hemoglobina i tjelesnoj masi bolesnika, kako je to prikazano u Tablici 6.6. Najčešća nuspojava je lokalizirani tromboflebitis, a najteža je već spomenuta anafilaktička reakcija zbog čega se bolesnike mora promatrati minimalno 30 minuta nakon primjene lijeka. Iako se na ovaj način postiže brži ispravak sideropenije, učinak na samu anemiju očekuje se, slično kao i kod peroralne nadoknade, tek tri do četiri tjedna od primjene.
|
Tablica 6.6. Parenteralna nadoknada željeza
|
|
Hemoglobin (g/L)
|
Tjelesna masa < 70 kg i doza Fe
|
Tjelesna masa > 70 kg i doza Fe
|
|
100 – 120 (žene)
|
1000 mg
|
1500 mg
|
|
100 – 130 (muškarci)
|
1000 mg
|
1500 mg
|
|
70 – 100 (žene i muškarci)
|
1500 mg
|
2000 mg
|
Ostale terapijske napomene. Transfuzija eritrocita indicirana je kod bolesnika s vrijednostima hemoglobina nižim od 70 g/L te ga u pravilu treba izbjegavati kad god je to moguće. Primjena jedne doze koncentrata eritrocita (cca 300 mL) povećava razinu hemoglobina za 8 do 10 g/L. Trudnicama, kao i ženama s izraženim ginekološkim krvarenjima (menoragija, metroragija), preporučena je profilaktička primjena željeza u dozi od 100 mg elementarnog željeza na dan.
Praćenje bolesnika. Krvnu sliku treba kontrolirati prvi put nakon dva tjedna od početka liječenja. Uspješan odgovor na terapiju podrazumijeva porast hematokrita na polovinu normalne vrijednosti nakon dva tjedna liječenja i normalnu vrijednost nakon dva mjeseca liječenja. Nakon korekcije anemije, krvnu se sliku kontrolira svaka tri mjeseca tijekom prve godine, a potom jednom godišnje..
SIDEROBLASTIČNA ANEMIJA
Definicija. Sideroblastična anemija predstavlja mikrocitnu ili makrocitnu hipokromnu anemiju nastalu uslijed poremećaja sinteze hema i posljedične nemogućnosti ugradnje željeza. Jedno od glavnih obilježja te vrste anemije pojava je prstenastih sideroblasta te povećana količina željeza u koštanoj sržim, što se dokazuje biopsijom koštane srži, dok se u laboratorijskim nalazima primjećuju znakovi preopterećenja željezom (povišene vrijednosti serumskoga željeza i feritina). Radi se o rijetkim oblicima anemije, a ovisno o nastanku razlikuju se nasljedni i stečeni oblik.
Nasljedna sideroblastična anemija nastaje uslijed genetski uvjetovanih poremećaja enzima koji sudjeluju u sintezi hema, a najčešće se radi o mutacijama gena za enzime - delta-aminolevulinsku sintetazu te hem-sintetazu. Ovi oblici sideroblastične anemije obično su povezani i s drugim genetskim poremećajima, pa često čine samo jedan dio ukupnoga kliničkog sindroma (npr. Pearsonov sindrom).
Stečena sideroblastična anemija može biti uzrokovana različitim bolestima i stanjima, kao što je alkoholizam, primjena nekih lijekova (kloramfenikol, izoniazid), manjak bakra i cinka, hipotermija i sl. Poseban oblik ove skupine anemija je refrakterna anemija s prstenastim sideroblastima, a predstavlja podoblik mijelodisplastičnoga sindroma.
Pristup bolesniku. Sumnju na sideroblastičnu anemiju pobuđuje nalaz mikrocitne hipokromne anemije uz povišene vrijednosti serumskog željeza i feritina (što je u suprotnosti sa sideropeničnom anemijom), a potvrđuje se biopsijom koštane srži. U uzorku koštane srži više je od 15 % prstenastih sideroblasta, a elektronskom mikroskopijom uočavaju se bogati depoziti željeza unutar mitohondrija. Liječenje ove skupine anemija je simptomatsko, a sastoji se od povremenih transfuzija eritrocita te zbrinjavanja povišene vrijednosti željeza u vidu venepunkcija ili primjene kelatora željeza (sprečavanje hemosideroze).
MEGALOBLASTIČNA ANEMIJA
Definicija. Megaloblastična anemija naziv je za anemiju koja nastaje kao posljedica poremećaja u sazrijevanju eritrocita uslijed nedostatka vitamina B12 ili folne kiseline (vitamin B9). Uslijed nedostatka tih vitamina dolazi do poremećaja u sintezi molekule DNA tijekom eritrocitopoeze, što rezultira nastankom megaloblasta u koštanoj srži, odnosno makrocita u perifernoj krvi.
Metabolizam vitamina B12. Vitamin B12 u organizam se unosi hranom životinjskoga porijekla (meso, riba, jaja, mlijeko), a predstavlja esencijalni element za organizam, s obzirom na to da sudjeluje kao koenzim u različitim biokemijskim procesima među kojima je i sinteza molekule DNA. U organizam se vitamin B12 unosi vezan za proteinski nosač koji se razgrađuje u želucu, a vitamin B12 potom se veže za kobalofilin, protein koji luče žlijezde slinovnice. Tako vezan, vitamin B12 dospijeva u dvanaesnik gdje dolazi do njegove disocijacije od kobalofilina da bi se potom vezao za unutarnji faktor (IF, engl. intrinsic factor) koji luče parijetalne stanice želučanoga epitela. Pomoću njega dolazi do ileuma u kojemu se vrši njegova internalizacija u enterocite u kompleksu s unutarnjim faktorom. U enterocitima dolazi do disocijacije kompleksa vitamin B12-unutarnji faktor te se vitamin B12 otpušta u cirkulaciju gdje se veže za transkobalamin II uz pomoć kojega se prenosi unutar organizma. Vitamin B12 neiskorišten u metaboličkim procesima pohranjuje se u hepatocite te jetra čini glavni izvor i skladište vitamina B12 u organizmu.
Metabolizam folne kiseline. Folna kiselina pripada skupini vitamina B (vitamin B9). Sudjeluje u različitim metaboličkim procesima, a ima i veliku ulogu tijekom diobe i diferencijacije stanice, osobito u embrionalnom periodu na način da djeluje kao enzim, koenzim i faktor rasta. Folna kiselina u organizam se unosi hranom biljnoga i životinjskog porijekla, obično u poliglutamatnom obliku. U gornjem se dijelu probavne cijevi poliglutamatni oblici folne kiseline razgrađuju u monoglutamatne oblike koji se potom preko enterocita apsorbiraju u cirkulaciju i dolaze do svojih ciljnih tkiva i stanica. Folna kiselina svoju ulogu na molekularnoj razini ostvaruje kao koenzim u procesima prijenosa atoma ugljika u metabolizmu aminokiselina i nukleinskih kiselina (osobito u procesima sinteze DNA i RNA).
Etiologija. Kako je navedeno, megaloblastična anemija nastaje uslijed nedostatka vitamina B12 i folne kiseline, na temelju čega se dijeli u tri skupine: megaloblastična anemija uslijed manjka vitamina B12, megaloblastična anemija uslijed manjka folne kiseline te megaloblastična anemija rezistentna na terapiju vitaminom B12 i folnom kiselinom (Tablica 6.7.).
Jedan od najčešćih uzroka nastanka megaloblastične anemije je manjak unutarnjeg faktora, kao posljedice kroničnoga autoimunog atrofičnog gastritisa. Takav oblik anemije naziva se perniciozna anemija. Oštećenje parijetalnih želučanih stanica rezultira smanjenim stvaranjem unutarnjega faktora zbog čega dolazi do smanjenje sposobnosti apsorpcije vitamina B12 i posljedičnoga razvoja anemije. Kronični atrofični gastritis predstavlja autoimuno oboljenje koje ima sljedeće karakteristike: 1) povezanost s prisutnošću određenih HLA antigena (HLA-B7, HLA-Dw2, HLA-Dw5,HLA-DR2), 2) prisutnost autoantitijela na antigen citoplazme parijetalnih stanica i/ili unutarnji faktor te 3) povezanost i povećana učestalost drugih autoimunih oboljenja.
|
Tablica 6.7. Etiologija megaloblastične anemije
|
|
Megaloblastična anemija uslijed manjka vitamina B12
|
Megaloblastična anemija uslijed manjka folne kiseline
|
Megloblastična anemija rezistentna na terapiju vitaminima B12 i B9
|
|
Nedostatan unos hranom;
Poremećaj apsorpcije:
- manjak unutarnjeg faktora,
- malapsorpcijski sindrom,
- resekcija ileuma,
crijevni paraziti;
Povećano iskorištavanje:
- trudnoća,
- hipertireoza,
- maligna bolest.
|
Nedostatan unos hranom;
Poremećaj apsorpcije:
- malapsorpcijski sindrom,
- lijekovi (antikonvulzivi, estrogeni);
Povećani gubitak:
- hemodijaliza;
Povećano iskorištavanje:
- trudnoća,
- hipertireoza,
- maligna bolest.
|
Prisutnost metaboličkih inhibitora:
- fluorouracil,
- citozin,
- azatiprin,
- merkaptopurin;
Poremećaji metabolizma folata;
Neobjašnjeni poremećaji.
|
Patogeneza. Manjak vitamina B12 i folne kiseline dovodi do poremećaja stvaranja molekule DNA u stanicama eritroidne loze što rezultira usporenim sazrijevanjem jezgre uz uredno sazrijevanje citoplazme (nukleo-citoplazmatska disocijacija). Posljedično izostaje smanjenje i izbacivanje jezgre iz eritrocita zbog čega oni postaju voluminozniji i sadržavaju veću količinu željeza u sebi (makrocitna hiperkromna anemija).
Klinička slika. Bolest se postupno razvija, a simptomi i znakovi ponajprije ovise o težini anemije. Najranije manifestacije uključuju osjećaj nedostatka zraka, palpitacije, vrtoglavicu, zujanje u ušima te glavobolju. Karakteristični trijas simptoma koji se viđa u bolesnika s pernicioznom anemijom je slabost, pečenje jezika te trnci (parestezije) u udovima. Nerijetko uslijed deficita vitamina B skupina nalazimo i druge specifične manifestacije (probavne i neurološke simptome). Probavni simptomi uključuju proljev, povraćanje i gubitak teka, dok neurološki uključuju pojavu simetričnih parestezija, gubitak osjeta vibracije, spastičnu ataksiju i sl. Pri kliničkom se pregledu zapaža blijedožuta boja kože i sluznica te karakteristične promjene na jeziku (jezik je gladak i crven, izraženih atrofičnih papila).
Dijagnoza megaloblastične anemije postavlja se na temelju laboratorijskoga nalaza koji uključuje snižene vrijednosti eritrocita i hemoglobina uz povišene vrijednosti eritrocitnih indeksa MCV-a i MCH-a (makrocitna hiperkromna anemija). Ovaj laboratorijski nalaz upućuje na manjak vitamina B12 i/ili folne kiseline kao mogućeg uzroka, stoga je potrebno odrediti njihove serumske vrijednosti (moguće je i odrediti vrijednosti folne kiseline u eritrocitima). Karakteristično, kod deficita vitamina B12 vrijednosti folne kiseline u serumu su normalne ili povišene, dok su kod deficita folne kiseline vrijednosti vitamina B12 umjereno snižene. Ostali laboratorijski nalazi uključuju umjereno snižene vrijednosti retikulocita, pojavu makroovalocita u razmazu periferne krvi te umjerenu trombocitopeniju i/ili leukopeniju i umjereno povišene vrijednosti laktat-dehidrogenaze i bilirubina kao odraza intramedularne destrukcije abnormlanih stanica eritroidne loze. Biopsija koštane srži rijetko se provodi, a ukazuje na normalnu morfologiju uz pojavu megaloblasta i kompenzatorne hiperplazije stanica eritroidne loze kao odgovora na neučinkovitu eritrocitopezu i manjak eritrocita. Zbog visoke učestalosti perniciozne anemije kod svih takvih bolesnika potrebno je obaviti gastroskopiju s biopsijom želuca te odrediti prisutnost autoantitijela na parijetalne stanice i unutarnji faktor.
Liječenje megaloblastične anemije podrazumijeva nadomjesno liječenje, a preporuča se istodobna primjena i vitamina B12 i folne kiseline.
Nadomjesno liječenje vitaminom B12 u prvom redu podrazumijeva njegovu parenteralnu primjenu (intramusukularno, subkutano) u obliku hidroksikobalamina u dozi od 1000 mcg svaki dan kroz prvi tjedan, potom 1000 mcg svaki tjedan tijekom prvog mjeseca, a potom 1000 mcg jednom mjesečno. Kod bolesnika s pernicioznom anemijom, gastrektomijom ili resekcijom ileuma ta se nadoknada vrši doživotno. Peroralna primjena vitamina B12 rijetko je indicirana i to većinom sa svrhom prevencije ponovnog razvoja deficita vitamina B12 nakon provedenoga parenteralnog liječenja. U tu se svrhu najčešće primjenjuje metilkobalamin u dozi od 1 mg/dan, a smatra se da to može biti dostatno i za bolesnike s pernicioznom anemijom jer se oko 1 % primijenjene doze apsorbira pasivnom difuzijom.
Nadomjesno liječenje folnom kiselinom primjenjuje se peroralnim pripravcima u dozi od 5 mg/dan kroz tri do četiri mjeseca, a profilaktički ga moraju uzimati trudnice i bolesnici na programu kronične hemodijalize. Oporavak i porast vrijednosti hemoglobina očekuje se za dva do tri tjedna liječenja, iako bolesnici navode i ranije simptomatsko olakšanje.
NASLJEDNE HEMOLITIČKE ANEMIJE
Uvod. Hemolitičke anemije predstavljaju skupinu anemija koje nastaju kao posljedica skraćenoga životnog vijeka i/ili prekomjerne razgradnje eritrocita. Prema etiologiji se razlikuju nasljedne i stečene hemolitičke anemije, a ovisno o mehanizmu nastanka razlikuju se prema tome jesu li nastale intravaskularnom ili ekstravaskularnom hemolizom. Glavne posljedice hemolitičkih anemija uključuju pojačanu produkciju eritropoetina, hiperplaziju stanica crvene loze u koštanoj srži, povećan broj retikulocita u perifernoj krvi, hiperbilirubinemiju te hemosiderozu.
Laboratorijski znakovi hemolize dijele se u dvije skupine, na pokazatelje razgradnje eritrocita te pokazatelje povećane kompenzacijske proizvodnje eritrocita. Pokazatelji razgradnje eritrocita su sniženje haptoglobina u serumu (najosjetljiviji laboratorijski pokazatelj hemolize), povećanje vrijednosti nekonjugiranoga bilirubina u serumu, povećanje vrijednosti urobilinogena u urinu, povišene vrijednosti laktat-dehidrogenaze u serumu te pojava fragmenata eritrocita u razmazu periferne krvi (shizociti). Pokazatelji povećane sinteze eritrocita su retikulocitoza i hiperplazija koštane srži.
Podjela. Nasljedne hemolitičke anemije posljedica su nasljednih poremećaja eritrocita koji dovode do skraćenja njegova životnog vijeka. Oni uključuju poremećaje membrane eritrocita, poremećaje metabolizma eritrocita te poremećaje sinteze globinskih lanaca (Tablica 6.8.).
|
Tablica 6.8. Najčešći oblici nasljednih hemolitičkih anemija
|
|
Poremećaji membrane eritrocita
|
Poremećaji metabolizma eritrocita
|
Poremećaji sinteze globinskih lanaca
|
|
Hereditarna sferocitoza
Hereditarna eliptocitoza
Hereditarna stomatocitoza
|
Deficit glukoza-6-fosfat dehidrogenaze
Deficit piruvat-kinaze
|
Alfa-talasemije
Beta-talasemije
Anemija srpastih stanica
|
HEREDITARNA SFEROCITOZA
Definicija. Hereditarna sferocitoza predstavlja najčešći oblik nasljedne hemolitičke anemije karakteriziran poremećajem građe citoskeleta membrane eritrocita zbog kojeg on poprima karakterističan sferičan (okrugao) izgled i postaje podložan sekvestraciji u slezeni. Procijenjena učestalost iznosi 1:5000.
Etiopatogeneza. Radi se o nasljednoj bolesti karakteriziranoj pojavom mutacije za neki od ključnih citoskeletnih proteina membrane eritrocita: spektrin (α i β), ankirin, vezni protein 3 te protein 4.2. Pretpostavljeni način nasljeđivanja je autosomno-dominantni, ali u oko 25 % slučajeva bolest se javlja bez pozitivne obiteljske anamneze, što ukazuje na nastanak de novo mutacije. U kliničkoj praksi najčešće se radi o poremećaju β-spektrina i ankrina. Posljedično manjku ili abnormalnoj strukturi nekoga od navedenih citoskeletnih proteina, membrana eritrocita ne može održavati svoj normalan bikonkavan oblik, nego dolazi do njezinoga izbočenja, pri čemu nastaju veliki i okrugli eritrociti. Takvi eritrociti ne mogu prilagođavati svoj oblik prilikom prolaska kroz kapilare slezene zbog čega bivaju zarobljeni i razgrađeni. Težina tako nastale hemolize uvelike varira između bolesnika s istim genetskim poremećajem. Posljedično povećanoj razgradnji eritrocita dolazi do razvoja splenomegalije (slezena može težiti i preko 1000 g), a razvija se i hiperbilirubinemija te s njom povezani kolelitijaza i kolecistitis. Iako je tijek bolesti obilježen različito izraženom hemolizom, povremeno su moguće i tzv. hemolitičke krize praćene pogoršanjem hemolize, najčešće su potaknute infekcijama, a mogu biti praćene i blokadom eritropoeze u koštanoj srži (aplastična kriza).
Klinička slika. Težina anemije različita je kod različitih bolesnika, a načelno se razlikuju tri klinička oblika bolesti: blagi, umjereni i teški oblik, čija su obilježja navedena u Tablici 6.9. Uz klasične simptome anemije (umor, malaksalost, glavobolja, nedostatak zraka) pri fizikalnom pregledu nalazimo različito izražen ikterus te splenomegaliju. Posljedično hiperbilirubinemiji bolesnici su skloniji razvoju kolelitijaze i kolecistitisa.
|
Tablica 6.9. Klinički oblici hereditarne sferocitoze
|
|
Blagi oblik
|
Umjereni oblik
|
Teški oblik
|
|
20 – 30 % slučajeva
Anemija i retikulocitoza odsutni.
Naznačena splenomegalija i ikterus
Bez potrebe za transfuzijom
|
60 – 75 % bolesnika
Anemija i značajna retikulocitoza
Jasna splenomegalija i ikterus
Povremene potrebe za transfuzijom
|
Oko 5 % bolesnika
Teška anemija i retikulocitoza
Izražena splenomegalija i ikterus
Trajna potreba za transfuzijom
|
Dijagnoza hereditarne sferocitoze zasniva se na anamnestičkim podacima (prisutnost bolesti u obitelji), fizikalnom pregledu (splenomegalija, ikterus) te laboratorijskim nalazima. U laboratorijskim nalazima nalazimo različito izraženu anemiju, retikulocitozu i hiperbilirubinemiju, a karakterističan je nalaz sferocita u razmazima periferne krvi, što je dovoljno za postavljanje dijagnoze. U svrhu potvrde dijagnoze mogu se provesti i genetska ispitivanja (ANK1, AE1, SPTB, SPTA1, EPB42 geni) te test osmotičke fragilnosti eritrocita (sferociti su izrazito osjetljivi na nagle osmotske promjene što se manifestira spontanom lizom pri izlaganju hiper- ili hipotonim otopinama).
Liječenje. Osnovu liječenja čini splenektomija kojom se produljuje životni vijek eritrocita i sprečava hemoliza. Izvodi se kod bolesnika s jasnom izraženim simptomima. Simptomatske transfuzije koncentriranih eritrocita provode se kod hemoglobina sniženoga ispod vrijednosti od 70 g/L. Kod težih se oblika preporuča i primjena vitamina B skupine (osobito folne kiseline). Prognoza splenektomiranih bolesnika načelno je dobra.
DEFICIT GLUKOZA-6-FOSFAT DEHIDROGENAZE
Definicija. Deficit enzima glukoza-6-fosfat dehidrogenaze najčešći je nasljedni oblik hemolitičke anemije koji nastaje uslijed poremećaja metabolizma eritrocita. Radi se o enzimopatiji koja rezultira povećanom razgradnjom eritrocita uslijed pojačane osjetljivosti starijih eritrocita na oksidativni stres ili ozljedu. Smatra se da više od 10 % ukupne populacije ima deficit tog enzima.
Etiopatogeneza. Glukoza-6-fosfat dehidrogenaza predstavlja enzim koji reducira NADP u NADPH koji djeluje kao antioksidans i štiti eritrocit od oksidativnoga stresa i učinka slobodnih kisikovih radikala. Gen koji kodira taj enzim nalazi se na X kromosomu pa je i njegovo nasljeđivanje vezano uz njega (X-vezano nasljeđivanje), što znači da bolest obično pogađa muškarce i rijetke homozigotne žene. Također, poznato je i više normalnih genskih inačica navedenoga gena, a najčešći je tip A (G6PD A+). Posljedično manjku ili potpunome nedostatku toga enzima i smanjenom stvaranju NADPH-a, eritrociti postaju osjetljivi na oksidativni stres i ozljede, zbog čega se prijevremeno uništavaju. To je posebno izraženo u slučaju starijih eritrocita, dok kod mlađih aktivnost enzima može biti i očuvana. Najčešći čimbenici koji predisponiraju oksidativno oštećenje eritrocita i njihovu razgradnju su lijekovi (Tablica 6.10.) te različite kemijske i biološke tvari (anilinske boje, naftalen, metilensko modrilo, bob – Vicia faba). Oštećenje eritrocita rezultira oksidacijom, denaturacijom i precipitacijom hemoglobina u eritrocitu u obliku Heinzovih tjelešaca koje makrofazi nastoje odstraniti pri prolasku kroz slezenu tako da nastaju specifični oblici eritrocita - bite-cells ili stanice kojima je «odgrižen» jedan dio membrane.
Klinička slika. Ovisno o težini poremećaja bolest se može prezentirati kao akutna intermitentna hemoliza ili kao kronična hemolitička anemija. Akutna intermitentna hemoliza obično je jače izražena, javlja se naglo i potaknuta je određenim stresorom (infekcija, primjena lijekova, izlaganje određenim kemikalijama) te je praćena klasičnim znakovima hemolitičke anemije. Drugi dio bolesnika obilježen je kroničnom hemolitičkom anemijom koja je stalno prisutna bez obzira na izlaganje nekim stresorima, a obično je blaže izražena i dobro se podnosi.
|
Tablica 6.10. Lijekovi koji izazivaju hemolizu u bolesnika s deficitom glukoza-6-fosfat dehidrogenaze
|
|
Antimalarici
|
Antibiotici
|
Ostali
|
|
Primakvin
Kinin
Pirimetamin
Klorokin
|
Sulfonamidi
Nitrofurantion
Klormafenikol
Fluorokinoloni
|
Aspirin
Vitamin K
Probenecid,
Fenilhidrazin
|
Dijagnoza se postavlja na temelju anamnestičkih podataka, fizikalnoga nalaza te laboratorijskih nalaza. U laboratorijskim nalazima prisutne su različito izražena anemija, retikulocitoza i hiperbilirbinemija (klasični nalazi kod hemolize), a najvažniji dijagnostički nalaz je pojava bite-cells eritrocita u razmazu periferne krvi. Dijagnoza se potvrđuje određivanjem vrijednosti (aktivnosti) enzima glukoza-6-fosfat dehidrogenaze.
Liječenje bolesnika je simptomatsko, povremenim transfuzijama koncentriranih eritrocita u slučaju pada vrijednosti hemoglobina ispod 70 g/L. Svim bolesnicima preporuča se izbjegavati uzimanje lijekova koji mogu precipitirati hemolizu, kao i drugih potencijalnih okidača hemolize.
TALASEMIJE
Uvod. Talasemije predstavljaju heterogenu skupinu genetskih poremećaja obilježenu smanjenom sintezom alfa ili beta-globulinskih lanaca tako da razlikujemo dvije temeljne skupine: alfa i beta-talasemije. Naziv 'talasemija' potječe iz vremena početka istraživanja ovakvih poremećaja koji su najviše bili zastupljeni kod osoba iz mediteranskoga područja (grč. thalassa - more). Genetski poremećaji koji rezultiraju razvojem talasemije u prvom se redu nasljeđuju autosomno-recesivnim putem, a opisani su i poremećaji koji se nasljeđuju autosomno-dominantno.
Alfa-talasemije nastaju kao posljedica poremećaja u sintezi α-globulinskih lanaca hemoglobina koji nastaje mutacijom jednoga ili sva četiri gena odgovorna za njihovu sintezu smještena na 16. kromosomu. Ovisno o težini poremećaja razlikuju se alfa+ talasemija, obilježena smanjenom sintezom, te alfa0 talasemija, obilježena potpunim izostankom sinteze α-globulinskih lanaca. Posljedično dolazi do kompenzacijskoga stvaranja veće količine β-globulinskih lanaca, tako da u konačnici nastaje hemoglobin koji se naziva hemoglobin H, koji se precipitira u starijim eritrocitima i dovodi do njihove hemolize.
Klinička slika ovisi o broju zahvaćenih gena, tako da postoje četiri klinička oblika: 1) fetalni hidrops (delecija sva četiri gena koja rezultira intrauterinom masivnom hemolizom i smrću ploda); 2) hemoglobinopatija H (delecija tri gena praćena minimalnom sintezom α-globulinskih lanaca, dovodi do težega oblika hemolitičke anemije); 3) alfa-talasemija minor (delecija dva gena, rezultira blagom i umjerenom hemolizom) te 4) stanje nosioca (delecija jednoga gena, asimptomatsko stanje).
Beta-talasemije nastaju kao posljedica poremećaja u sintezi β-globulinskih lanaca uslijed mutacija gena koji se nalazi na 11. kromosomu, a do danas je opisano više od 200 mutacija s različitim fenotipskim posljedicama. Kao i kod alfa-talasemija, razlikuju se dva oblika ovisno o težini poremećaja: beta+ talasemija, praćena smanjenom sintezom, te beta0 talasemija, praćena potpunim izostankom sinteze β-globulinskih lanaca. U beta-talasemiji javlja se višak α-globulinskih lanaca koji precipitiraju i dovode de hemolize eritrocita.
Tri su glavna klinička oblika beta-talasemija: beta-talasemija major, beta-talasemija minor te beta-talasemija intermedija. Beta-talasemija major javlja se kod homozigota, već u djetinjstvu, a obilježena je teškim oblikom hemolitičke anemije koja zahtijeva stalne povremene transfuzije koncentriranih eritrocita te uzrokuje hiperbilirubinemije, splenomegalije i sekundarne hemokromatoze (nakupljanje željeza uslijed hemolize i čestih transfuzija). Beta-talasemija intermedija može se javiti i kod homozigota i heterozigota, a obilježena je srednje izraženom hemolitičkom anemijom koja ne zahtijeva česte transfuzije eritrocita. Beta-talasemija minor javlja se kod heterozigota i praćena je blagom asimptomatskom anemijom te ne zahtijeva transfuzije eritrocita.
Dijagnoza talasemija temelji se na anamnestičkim podacima (prisutnost anemije u obitelji), fizikalnom pregledu te laboratorijskim nalazima u kojima nalazimo mikrocitnu, hipokromnu anemiju praćenu retikulocitozom i nalazom target-cell (stanice poput cilja) u razmazu periferne krvi. Metodom elektroforeze ili tekućinske kromatografije moguće je odijeliti i uočiti patološke oblike hemoglobina (npr. hemoglobin H). Ključan korak u postavljanju dijagnoze talasemija je gensko testiranje i određivanje genetskih promjena.
Liječenje bolesnika je simptomatsko i ovisi o težini bolesti. Primjenjuju se transfuzije koncentriranih eritroicta kako bi se održala zadovoljavajuća vrijednost hemoglobina, kelatori željeza kako bi se spriječio nastanak sekundarne hemokromatoze te folna kiselina. Splenektomija može privremeno dovesti do poboljšanja i smanjenja potrebe za učestalim transfuzijama. U najtežim oblicima indicirana je i transplantacija alogenih matičnih stanica koja ima povoljan učinak u više od 90 % bolesnika.
ANEMIJA SRPASTIH STANICA
Definicija. Anemija srpastih stanica označava nasljednu hemolitičku anemiju obilježenu stvaranjem patološkoga hemoglobina S zbog čega eritrociti poprimaju karakterističan oblik nalik srpu (srpasti eritrociti), što uzrokuje njihovu jaču sklonost hemolizi i stvaranju vaskularnih okluzija.
Etiopatogeneza. Radi se o nasljednoj bolesti koja nastaje uslijed točkaste mutacije gena za beta-globinski lanac hemoglobina (zamjena glutamina valinom na 6. poziciji), zbog čega se stvara patološka forma hemoglobina koja se naziva hemoglobin S. Eritrociti koji sadrže hemoglobin S u uvjetima hipoksije, hiperkapnije ili hipertermije mijenjaju svoj oblik, pri čemu poprimaju srpasti izgled (drepanociti) i zbog toga su podložni sekvestraciji u slezeni, kao i otežanom prolazu kroz kapilare pri čemu stvaraju vaskularne okluzije koje mogu rezultirati ishemičkim oštećenjima (mikroinfarkti mozga, bubrega, jetre, pluća).
Klinička slika. Anemija srpastih stanica očituje se teškom hemolitičkom anemijom, okluzivnim vaskularnim komplikacijama te kroničnom hiperbilirubinemijom. Kliničke manifestacije obično su potaknute nekim okidačem, najčešće infekcijama ili sistemskom hipoksijom uslijed respiratornih poremećaja. S obzirom da ishemijom mogu biti zahvaćeni gotovo svi organi, klinička slika može biti šarolika: akutno bubrežno oštećenje, poremećaji stanja svijesti, crijevna ishemija, jetrena insuficijencija i sl.
Dijagnoza se postavlja na osnovi kliničke slike, anamnestičkih podataka (obiteljska pojavnost bolesti) te laboratorijskih nalaza koji uključuju anemiju, retikulocitozu, hiperbilirubinemiju i nalaz srpastih eritrocita u razmazu periferne krvi. Elektroforeza može detektirati prisutnost hemoglobina S. Konačna dijagnoza postavlja se na temelju dokaza genetske mutacije.
Liječenje je simptomatsko i u prvom redu uključuje primjenu transfuzija koncentrata eritrocita uz provođenje mjera sprečavanja razvoja sekundarne hemokromatoze. Vaskularne krize vezane uz srpastu anemiju liječe se obilnom hidracijom, primjenom antibiotika, analgetika i ostalim mjerama simptomatskoga liječenja. Alogena transplantacija matičnih stanica može biti indicirana kod najtežih oblika bolesti. Unatoč mjerama liječenja, prognoza je loša. Oko 15 % bolesnika s tom anemijom umre u prva dva desetljeća života, a oko 50 % u dobi od četrdeset godina.
STEČENE HEMOLITIČKE ANEMIJE
Uvod. Stečene hemolitičke anemije predstavljaju skupinu hemolitičkih anemija koje nastaju djelovanjem vanjskih čimbenika na eritrocite, bilo da se radi o imunosnim bilo o neimunosnim čimbenicima, pa se prema tome i dijele na imunosne hemolitičke anemije i neimunosne hemolitičke anemije. S obzirom na to da životni vijek eritrocita nije skraćen uslijed poremećaja strukture eritrocita, često se ova skupina anemija naziva i ekstrakorpuskularnim anemijama.
IMUNOSNE HEMOLITIČKE ANEMIJE
Definicija. Imunosne hemolitičke anemije su anemije kod kojih je skraćeni životni vijek eritrocita, odnosno njihova destrukcija posredovana tzv. antieritrocitnim antitijelima. Ako se radi o antieritrocitnim antitijelima usmjerenima prema vlastitim antigenima, onda govorimo o autoimunoj hemolitičkoj anemiji, a kada se radi o antieritrocitnim antitijelima usmjerenima protiv nevlastitih antigena, govorimo o aloimunoj hemolitičkoj anemiji (npr. hemolitička reakcija nakon primjene transfuzije eritrocita krive krvne grupe). Osnovna laboratorijska karakteristika imunosnih hemolitičkih anemija pozitivan je Coombsov test.
Coombsovim testom dokazuje se prisutnost antieritrocitnih antitijela, a ovisno o načinu njegova izvođenja razlikuju se direktni i indirektni Coombsov test. Test se temelji na procesu aglutinacije eritrocita, što označava sakupljanje i sljepljivanje eritrocita posredovano reakcijom antigen-antitijelo. U fiziološkim okvirima površina eritrocita je negativna i oni se međusobno odbijaju što onemogućava njihovo međusobno sljepljivanje (aglutinaciju). Ukoliko se na membrani eritrocita pojave antitijela vezana na specifične površinske antigene ili komponentne komplementa (C3 komponenta), što je slučaj kod imunosnih hemolitičkih anemija, moguće je primjenom životinjskog protutijela (anti-IgG, anti-C3d) postići njihovu aglutinaciju. Ona se ostvaruje na način da se primijenjena antitijela jednim dijelom vežu za antieritrocitno protutijelo ili C3 komponentu jednoga eritrocita, a drugim dijelom za antieritrocitno protutijelo ili C3 komponentu drugoga eritrocita te tako posreduju njihovo međusobno spajanje tj. aglutinaciju.
Direktni Coombsov test koristi se za izravno dokazivanje prisutnosti antieritrocitnih antitijela ili komponenata komplementa (C3 komponenta) na površini eritrocita, kao glavne odlike imunosnih hemolitičkih anemija. Test se sastoji od ispiranja eritrocita fiziološkom otopinom kako bi na njima ostala samo antitijela i komponente komplementa vezane za njihovu površinu. Dodavanjem životinjskoga seruma koji sadrži anti-IgG ili anti-C3 dolazi do agultinacije eritrocita prema prethodno opisanome principu – ako se na membranama eritrocita nalaze antieritrocitna protutijela ili C3 komponenta komplementa.
Indirektni Coombsov test koristi se za neizravno dokazivanje prisutnosti antieritrocitnih antitijela u serumu bolesnika, a izvodi se tako da se najprije bolesnikov serum inkubira s normalnim eritrocitima, što dovodi do vezanja antieritrocitnih antitijela na dodane eritrocite (ako ona postoje) te se potom provede klasični Coombsov test (dodavanje anti-IgG) što će rezultirati nastankom aglutinacije i test označiti pozitivnim.
Pozitivnost i direktnoga i indirektnoga Coombsovog testa ukazuje na postojanje autoimune hemolitičke anemije, dok pozitivan indirektni uz negativan direktni Coombsov test uglavnom ukazuje na aloimunu hemolitičku anemiju.
AUTOIMUNE HEMOLITIČKE ANEMIJE
Definicija i podjela. Autoimune hemolitičke anemije predstavljaju grupu hemolitičkih anemija koje nastaju uslijed stvaranja autoantitijela na eritrocitne antigene zbog čega dolazi do njihove destrukcije i uništenja. Ovisno o temperaturi pri kojoj su ta autoantitijela aktivna, razlikujemo autoimunu hemolitičku anemiju posredovanu toplim i autoimunu hemolitičku anemiju posredovanu hladnim autoantijelima. Glavne razlike između ta dva oblika anemije prikazane su u Tablici 6.11.
Autoimuna hemolitička anemija posredovana toplim autoantitijelima ona je kod koje antieritrocitna antitijela pokazuju najveću reaktivnost pri tjelesnoj temperaturi od 37 ℃ i ona čini 80 % autoimunih hemolitičkih anemija. Najčešće se radi o antieritrocitnim antitijelima klase IgG, rijetko se radi o IgA ili IgM antitijelima usmjerenima na različite antigene na površini eritrocita, uključujući i Rh antigene. Uz njih se na površini eritrocita mogu nalaziti i komponente komplementa, najčešće C3 komponenta (Coombsov test pozitivan i na anti-IgG i na anti-C3).
Autoimuna hemolitička anemija posredovana hladnim autoantitijelima ona je kod koje antieritrocitna antitijela pokazuju najveću reaktivnost pri tjelesnoj temperaturi nižoj od 37 ℃, obično nižoj od 32 ℃. Ona čini oko 20 % autoimunih hemolitičkih anemija, a antieritrocitna antitijela uglavnom su IgM klase i usmjerena su na tzv. „I“ antigen eritrocitne membrane. I kod takvih se bolesnika uz autotoantitijela na površini eritrocita nalaze komponente komplementa, zbog čega je Coombsov test pozitivan samo na anti-C3.
|
Tablica 6.11. Karakteristike autoimunih hemolitičkih anemija
|
|
Osnovne karakteristike
|
Autoimuna hemolitička anemija posredovana toplim autoantitijelima
|
Autoimuna hemolitička anemija posredovana hladnim autoantitijelima
|
|
Učestalost
|
80 %
|
20 %
|
|
Antitijela
|
IgG klasa
|
IgM klasa
|
|
Antigeni
|
Antigeni Rh sustava
|
I antigen
|
|
Coombsov test
|
Anti-IgG pozitivan, Anti-C3 pozitivan
|
Anti-C3 pozitivan
|
Etiopatogeneza. S obzirom na uzrok, razlikujemo primarne i sekundarne autoimune hemolitičke anemije. Primarne označavaju autoimunu hemolitičku anemiju kao izoliranu bolest pri kojoj se stvaraju autoantitijela na antigene eritrocita, zbog nepoznatoga uzroka. Sekundarne autoimune hemolitičke anemije javljaju se u sklopu drugih, obično autoimunih, bolesti te je hemolitička anemija samo dio ukupne kliničke ekspresije bolesti. Najčešće se radi o limfoproliferativnim bolestima, autoimunim bolestima vezivnoga tkiva (reumatoidni artritis, sistemski eritematozni lupus), kroničnim infektivnim bolestima, solidnim neoplazmama ili lijekovima. Eritrociti obloženi autoantitijelima postaju podložni uništenju od strane stanice monocitno-makrofagnoga sustava (osobito u slezeni) ili su skloni pojačanoj razgradnji posredovanoj aktivacijom komplementa. Izravni citotoksični učinak senzibiliziranih eritrocita u nastanku takvih anemija nije do kraja razjašnjen
Klinička slika. Primarne autoimune hemolitičke anemije rjeđe se javljaju, obično se radi o sekundarnima kojima dominiraju simptomi i znaci osnovne bolesti, tako da se anemija uglavnom otkriva uz pomoć patoloških laboratorijskih nalaza. Anemija se uglavnom postupno razvija, iako postoje i brzoprogresivni oblici. Autoimuna hemolitička anemija posredovana toplim autoantitijelima može se kretati u rasponu od umjerene do teške, dok je autoimuna hemolitička anemija posredovana hladnim antitijelima uglavnom blaga i izraženija je u zimskim mjesecima. Simptomi i znakovi koji prate hemolitičke anemije uključuju umor, malaksalost, vrtoglavicu, zujanje u ušima i palpitacije, a karakteristični su i blago do umjereno izražena žutica te tamni urin kao posljedica hiperbilirubinemije.
Dijagnoza se postavlja na temelju laboratorijskih nalaza. Osim sniženih vrijednosti eritrocita i hemoglobina, karakteristični laboratorijski nalazi koji upućuju na hemolizu su retikulocitoza, povišene vrijednosti laktat dehidrogenaze, hiperbilirubinemija (indirektna) te snižene vrijednosti haptoglobina. Kod svih je bolesnika pozitivan direktan Coombsov test, a ako je količina antierirocitnih antitijela visoka, oni se nalaze i u slobodnom obliku u plazmi, pa je pozitivan i indirektan Coombsov test. Kod svih bolesnika potrebno je ovisno o anamnestičkim podacima i kliničkom nalazu provesti dodatnu dijagnostičku obradu s ciljem otkrivanja uzroka autoimune hemolitičke anemije.
Liječenje. U sekundarnim oblicima bolesti korekcija anemija postiže se liječenjem osnovne bolesti. Blagi do umjereni oblici anemije koji ne dovode do značajnih simptoma ne zahtijevaju posebne oblike liječenja. Transfuzija eritrocita primjenjuje se kada vrijednost hemoglobina padne ispod 70 g/L. Prvu liniju liječenja čini peroralna ili parenteralna primjena kortikosteroida i to metilprednizolona u dozi od 1 mg/kg/dan. Doziranje se prilagođava vrijednostima hemoglobina pri čemu se njegove vrijednosti nastoje održavati iznad 100 g/L. Nakon postupnoga sniženja doze, kortikosteroidi se ukidaju nakon tri ili četiri mjeseca remisije. Drugu liniju liječenja čine splenektomija ili primjena rituksimaba, a iznimno se primjenjuju i imunosupresivi (ciklosporin, azatioprin), intravenski imunoglobulini ili plazmafereza. Autoimuna hemolitička anemija posredovana toplim protutijelima pokazuje bolje rezultate pri liječenju nego autoimuna hemolitička anemija posredovana hladnim protutijelima.
Prognoza. Primarni oblici bolesti imaju varijabilan i nepredvidiv tijek bilježen čestim egzarcebacijama i remisijama, ali načelno je desetogodišnje preživljenje veće od 70 %. Prognoza sekundarnih oblika vezana je uz prognozu osnovne (uzročne) bolesti. Općenito, sekundarni su oblici obilježeni težim kliničkim tijekom.
ALOIMUNE HEMOLITIČKE ANEMIJE
Aloimune hemolitičke anemije posredovane su antieritrocitnim antitijelima koja nisu usmjerena na vlastite antigene. Glavni oblici takvih anemija su hemolitička bolest novorođenčadi te hemolitička reakcija nakon transfuzije eritrocita krive krvne grupe (najčešće se radi o AB0 inkompatibilnosti). Ako se u bolesnika infundiraju eritrociti koji imaju na svojoj površini antigene strane bolesniku, najprije dolazi do senzibilizacije, a potom i do prave hemolitičke reakcije.
NEIMUNOSNE HEMOLITIČKE ANEMIJE
Uvod. Neimunosne hemolitičke anemije su anemije kod kojih destrukcija i razgradnja eritrocita nisu uvjetovane imunološkom reakcijom. Imaju isti laboratorijski obrazac kao i imunosne hemolitičke anemije, jedina je razlika što kod neimunosnih Coombsov test nije pozitivan. Te vrste hemolitičkih anemija mogu biti uzrokovane biološkim, kemijskim ili fizikalnim čimbenicima.
Neimunosne hemolitičke anemije uzrokovane biološkim čimbenicima povezane su s razvojem različitih infekcija. U takvim stanjima hemolitička anemija može nastati kao posljedica izravnoga oštećenja eritrocita (malarija, bartoneloza) ili neizravnoga oštećenja (Escherichia coli, Clostridium velchi).
Neimunosne hemolitičke anemije uzrokovane kemijskim čimbenicima mogu biti posljedica unosa određenih lijekova ili toksina. Neki lijekovi, kao što je ribavarin, imaju sposobnost direktnoga citotoksičnog učinka na eritrocite, kao i neki toksini (olovo, arsen, bakar, formaldehid, zmijski otrov).
Neimunosne hemolitičke anemije uzrokovane fizikalnim čimbenicima predstavljaju najčešći oblik neimunosnih hemolitičkih anemija. One nastaju djelovanjem mehaničke sile (traume) na eritrocite, a ovisno o kliničkim okolnostima razlikujemo makroangiopatsku i mikroangiopatsku mehaničku hemolitičku anemiju.
Makroangiopatska mehanička hemolitička anemija najčešće je uzrokovana grubim intravaskularnim mehaničkim zaprekama kao što su arteriovenske anastomoze, defekti srčane pregrade, umjetne srčane valvule ili izraženim utjecajem vanjskih mehaničkih sila, kao što su dugotrajno marširanje (tzv. marš hemoglobinurija prisutna kod vojnika nakon dugotrajnoga hodanja), maratonske utrke, boks i sl. Splenomegalija također predstavlja jedan od uzroka makroangiopatske hemolitičke anemije (hipersplenizam).
Mikroangipatska mehanička hemolitička anemija nastaje uslijed intravaskularne hemolize koja nastaje fragmentacijom eritrocita pri prolasku kroz patološki promijenjene krvne žile manjega promjera (obično kapilare), a glavni primjeri takvih anemija su trombotička trombocitopenična purpusa, hemolitičko-uremični sindrom, hemoliza udružena s diseminiranom intravaskularnom koagulopatijom, hemoliza povezana s eklampsijom i preeklampsijom i sl.
POREMEĆAJI BROJA I FUNKCIJE TROMBOCITA
Najznačajniji poremećaj trombocita u kliničkoj praksi smanjenje je njihova broja, odnosno trombocitopenija, koja je izravno povezana s povećanim rizikom od nastanka krvarenja, a u nekim slučajevima i s pojavom venskih i/ili arterijskih tromboza. Povećan broj trombocita ili trombocitoza nešto se rjeđe susreće, najčešće u sklopu kroničnih mijeloproliferativnih bolesti (esencijalna trombocitoza) ili kao sekundarni poremećaj. Poremećaji funkcije trombocita također predstavljaju rijetku skupinu poremećaja koji se mogu javiti kongenitalno - u sklopu različitih nasljednih sindroma, ili stečeno - najčešće uslijed primjene različitih lijekova
TROMBOCITOPENIJA
Uvod. Trombocitopenija se definira smanjenim brojem trombocita, i to manjim od 150 x 109/L. Rizik od pojave krvarenja kod takvih bolesnika izravno je povezan s brojem trombocita (Tablica 6.12.). Trombocitopenija se može razviti na temelju tri osnovna patofiziološka mehanizma: smanjeno stvaranje i/ili pojačana destrukcija trombocita te pojačana sekvestracija u slezeni.
|
Tablica 6.12. Trombocitopenija i rizik od krvarenja
|
|
Broj trombocita
|
Rizik od krvarenja
|
|
> 100 x 109/L
|
Bez rizika (čak ni kod većih operacija)
|
|
50 – 100 x 109/L
|
Produljeno krvarenje vezano uz veće (rizičnije) zahvate i ozljede
|
|
20 – 50 x 109/L
|
Produljeno krvarenje može se javiti i pri manjim zahvatima i ozljedama
|
|
< 20 x 109/L
|
Mogućnost pojave spontanoga krvarenja
|
Pojačana destrukcija trombocita jedan je od najčešćih patofizioloških mehanizama nastanka trombocitopenije, a karakterizira ga uredno stvaranje trombocita u koštanoj srži te skraćeni životni vijek posljedično imunološkim ili neimunološkim reakcijama i mehanizmima. Najznačajniji poremećaji koji dovode do trombocitopenije neimunološkim mehanizmima jesu sepsa, diseminirana intravaskularna koagulopatija, hemolitičko-uremični sindrom, trombotička trombocitopenična purpura, preeklampsija i eklampsija, ekstrakorporalna cirkulacija, prisutnost velikih kavernoznih hemangioma i sl. Imunološki posredovana destrukcija trombocita viđa se u bolesnika s idiopatskom (imunom) trombocitopenijom te lijekovima uzrokovanoj trombocitopeniji. Nalaz biopsije koštane srži u ovih bolesnika je uredan, odnosno u nekih možemo naći i povećani broj megakariocita kao kompenzacijski odgovor na pojačani gubitak (destrukciju) trombocita u perifernoj krvi.
Pojačana sekvestracija trombocita također je čest uzrok stečene trombocitopenije, a susreće se kod bolesnika s povećanom slezenom u kojoj se i u normalnim okolnostima uklanja oko 30 % ukupne mase trombocita. Tako nastalu trombocitopeniju rijetko karakterizira smanjenje broja trombocita ispod 50 x 109/L, pa su i krvarenja povezana s njom rijetka.
Smanjeno stvaranje trombocita u koštanoj srži najrjeđi je uzrok nastanka trombocitopenije, a primarno ga obilježava gubitak ili smanjeni broj megakariocita iz kojih nastaju trombociti u koštanoj srži. Uzroci mogu biti različiti: poremećaj matične hematopoetske stanice (nasljedna ili stečena aplastična anemija), infiltracija koštane srži (leukemijske stanice, mijelom, limfom, granulomatoza), utjecaj kemoterapije i/ili radioterapije te nutritivni poremećaji (deficijencija folata i vitamina B12). Trombocitopenija se u takvim uvjetima rijetko pojavljuje samostalno, obično je dio pancitopenije zajedno s anemijom i leukopenijom.
Najznačajniji specifični oblici trombocitopenije su: idiopatska (imuna) trombocitopenija, lijekovima inducirana trombocitopenija uz trombocitopeniju induciranu heparinom kao izdvojeni klinički entitet, trombotička trombocitopenična purpura, hemolitičko-uremični sindrom te diseminirana intravaskularna koagulopatija. Svaki od navedenih poremećaja bit će opisan zasebno.
Transfuzija trombocita. S obzirom na to da je u većini ovih stanja temelj simptomatskoga liječenja transfuzija trombocita, potrebno je naglasiti nekoliko činjenica vezano uz njezinu primjenu. Trombocite koji se primjenjuju u liječenju ovih bolesnika dobiva se na dva načina: 1) uzimanjem trombocita iz donirane pune krvi, pri čemu se trombociti moraju pulirati iz krvi više davatelja (obično četiri do šest) kako bi se dobila jedna doza (tzv. pool) koncentriranih trombocita ili 2) postupkom afereze (trombofereza) pri čemu se od davatelja uzimaju samo trombociti, tako da dobivena doza trombocita potiče od samo jednoga davatelja, a taj postupak i znatno smanjuje mogućnost aloimunizacije primatelja (za razliku od klasičnoga uzorkovanja pri čemu se bolesniku daje doza trombocita sastavljena od populacije trombocita više davatelja). Jedna doza (pool) trombocita sadržava oko 50 bilijuna trombocita, a to je dostatno da bi se broj trombocita u perifernoj krvi povećao za 10 do 12 x 109/L. Specifično je za transfuziju trombocita njihovo skladištenje koje se mora vršiti na sobnoj temperaturi, s obzirom na to da su trombociti osjetljivi na niže temperature. Takav način pohrane povezan je s većim rizikom od nastanka bakterijske kontaminacije i prijenosa bakterijskih infekcija u odnosu na koncentrirane eritrocite i plazmu koji se zamrzavaju, a skraćen je i životni vijek trombocita, tako da ih se mora potrošiti unutar pet do sedam dana. Kod primjene trombocita u liječenju trombocitopenija dvije su osnovne vodilje: vrijednost trombocita i vrsta trombocitopenije. Primjena koncentrata trombocita može biti profilaktička i terapijska. Profilaktička primjena indicirana je kod svih bolesnika s brojem trombocita manjim od 5 do 10 x 109/L, kako bi se smanjio rizik od spontanoga krvarenja, te kod bolesnika kod kojih se planira određeni invazivni postupak ili zahvat - tada se primjena trombocita usklađuje s vrstom intervencije i potencijalnim rizikom od krvarenja (Tablica 6.13.). Terapijska primjena trombocita indicirana je kod bolesnika s krvarenjem pri čemu se njihov broj mora održavati većim od 50 x 109/L, odnosno 100 x 109/L ako se radi o krvarenju u središnji živčani sustav. Primjenu trombocita treba izbjegavati kod bolesnika s trombotičkom trombocitopeničnom purpurom, kao i kod bolesnika s trombocitopenijom induciranom heparinom zbog naravi bolesti (kasnije opisano), jer tada primjena trombocita može biti povezana s pogoršanjem same bolesti, odnosno progresijom ishemije uslijed arterijsko-venskih trombotičkih incidenata.
|
Tablica 6.13. Profilaktička primjena transfuzije trombocita
|
|
Rizik od krvarenja
|
Preporučen broj trombocita
|
Primjeri (vrste) zahvata
|
|
Visokorizični zahvati/intervencije
|
75 – 100 x109/L
|
Neurokirurški zahvati
Kirurgija oka
Kirurgija štitnjače i prostate
|
|
Srednjerizični zahvati/intervencije
|
50 x 109/L
|
Većina kirurških zahvata
Biopsija jetre
Ekstrakcija zuba, oralna kirurgija
|
|
Niskorizični zahvati/intervencije
|
20 – 30 x 109/L
|
Endoskopske procedure
Lumbalna punkcija
Središnji venski kateteri
|
IDIOPATSKA (IMUNA) TROMBOCITOPENIJA
Definicija i epidemiologija. Imuna trombocitopenija (ITP) predstavlja oblik trombocitopenije koji nastaje uslijed perzistiranja cirkulirajućih antitrombocitnih antitijela koja se vežu na površinu trombocita, zbog čega se oni pojačano uklanjaju putem stanica retikuloendotelnoga sustava. Bolest se javlja s učestalošću od 8 oboljelih na 100 000 stanovnika, češće kod mlađih žena, iako se može javiti i u drugim životnim dobima kod oba spola.
Etiopatogeneza. Idiopatska trombocitopenija može se javiti u dva oblika: primarni i sekundarni. Primarna idiopatska trombocitopenija nepoznatoga je uzroka, a javlja se kao izolirana bolest, za razliku od sekundarne koja se javlja u sklopu drugih bolesti, kao što su sistemske bolesti vezivnoga tkiva (sistemski eritematozni lupus), limfoproliferativne bolesti, ili može biti potaknuta lijekovima ili infekcijama (hepatitis C infekcija, HIV infekcija, Helicobacter pylori infekcija). Antitrombocitna antitijela najčešće su usmjerena na glikoproteine koje nalazimo na površini trombocita (receptori IIb/IIIa i Ib/IX), a smatra se da mogu imati utjecaj i na megakariocite u koštanoj srži, s obzirom na prateći neodgovarajući odgovor koštane srži u pogledu povećanoga (kompenzatornog) odgovora na smanjenje broja trombocita.
Klinička slika. U odrasloj se dobi imuna trombocitopenija najčešće manifestira kao kronična i recidivirajuća bolest, za razliku od one u pedijatrijskoj populaciji gdje se obično prezentira akutnim obilježjima. Najčešće kliničke manifestacije ovoga poremećaja uključuju difuzna kožna krvarenja (sufuzije, ekhimoze, petehije, hematomi), iako se mogu javiti i druge manifestacije hemoragijske dijateze: epistaksa, krvarenja iz gingive, hemoptize, hematurija, melena i sl. Kod žena su česta produljena i obilna menstruacijska krvarenja. Spontana krvarenja obično se javljaju kod bolesnika s ukupnim brojem trombocita manjim od 10 x 109/L.
Dijagnoza se postavlja na osnovi laboratorijskih nalaza u kojima dominira izolirana trombocitopenija uz moguću mikrocitnu anemiju ako bolesnik ima manifestno krvarenje. Biopsija koštane srži indicirana je kod bolesnika koji imaju neobjašnjenu citopeniju koja zahvaća dvije ili tri loze, kod bolesnika s izoliranom trombocitopenijom koji su stariji od 40 godina te kod bolesnika s izoliranom trombocitopenijom koji ne reagiraju na standardno liječenje idiopatske trombocitopenije. Koštana srž takvih bolesnika u pravilu je uredna ili pokazuje povećani broj megakariocita (kompenzatorni odgovor). Kod dvije trećine bolesnika moguće je dokazati antitrombocitna antitijela u serumu. Svim bolesnicima s izoliranom trombocitopenijom potrebno je napraviti serološku dijagnostiku HCV i HIV infekcije, kao i testiranje na Helicobacter pylori infekciju.
Liječenje se provodi kod bolesnika koji imaju broj trombocita niži od 20 x 109/L te bolesnika s aktivnim krvarenjem. Inicijalno liječenje podrazumijeva primjenu kortikosteroida (prednizolon 1 mg/kg/dan per os kroz sedam do deset dana s postupnim snižavanjem ili visoke doze deksametazona od 40 mg/dan intravenski kroz četiri dana svakih 28 dana kroz četiri do šest ciklusa) s ili bez primjene intravenskih imunoglobulina (1 g/kg/dan intravenski kroz jedan do dva dana) ili anti-D imunoglobulina (75 mcg/kg jednokratno intravenski). Odgovor na primjenu kortikosteroida očekuje se kroz pet do sedam dana, a kod istodobne primjene imunoglobulina i ranije (dva do tri dana). Kod bolesnika s manifestnim krvarenjem i trombocitopenijom primjenjuju se i transfuzije trombocita. Kod bolesnika koji ne pokazuju odgovor na ovo liječenje prelazi se na drugu liniju liječenja. Drugu liniju liječenja prvenstveno predstavlja primjena monoklonalnoga anti-CD20 antitijela (rituksimab) u dozi od 375 mg/m2/tjedno intravenski kroz četiri tjedna, a u obzir dolazi i promjena intravenskih imunoglobulina, anti-D imunoglobulina, splenektomija te agonisti trombopoetinskih receptora (romiplostim, eltrombopag). Ukoliko ni na te lijekove bolesnici ne pokazuju odgovor, može se pokušati i s primjenom ciklosporina, azatioprina ili mikofenolata.
Prognoza takvih bolesnika načelno je dobra. Velik dio bolesnika ima asimptomatsku izoliranu trombocitopeniju, a kod simptomatskih se bolesnika u više od dvije trećine postiže dobar klinički odgovor na standardno (inicijalno) liječenje.
TROMBOTIČKA TROMBOCITOPENIČNA PURPURA
Definicija i epidemiologija. Trombotička trombocitopenična purpura (TTP) ili Morbus Moskowitz predstavlja oblik potrošne trombocitopenije udružene s hemolitičkom anemijom koja nastaje uslijed izostanka cijepanja ultravelikih multimera von Willenbrandova faktora zbog stečenoga ili nasljedno uvjetovanoga manjka ADAMTS13 metaloproteinaze koja posreduje ovu reakciju cijepanja. Radi se o rijetkoj, ali potencijalno po život opasnoj bolesti ukoliko ju se na vrijeme ne prepozna i ne započne s njezinim liječenjem. Incidencija bolesti iznosi 3,7 oboljelih na 100 000 stanovnika, a najčešće vrijeme manifestacije je treće i četvrto desetljeće života.
Etiopatogeneza. Osnovni poremećaj u TTP-u predstavlja manjak ADAMTS13 metaloproteinaza koja posreduju cijepanje ultravelikih multimera von Willenbrandova faktora što ima veliku ulogu u aktivaciji trombocita. Von Willenbrandov faktor luče endotelne stanice u obliku ultravelikih multimera koji ostvaruju jak učinak na aktivaciju trombocita, a djelovanjem ADAMTS13 metaloproteinaza razgrađuju se na manje segmente koji nemaju toliko izražen protrombocitni učinak. Deficit ADAMTS13 metaloproteinaza može se javiti kao kongenitalni (mutacija ADAMTS13 gena) ili kao stečeni poremećaj (idiopatsko stvaranje IgG autoantitijela na molekulu ADAMTS13). Posljedično navedenom deficitu i nakupljanju ultravelikih multimera von Willenbrandova faktora pojačano se aktiviraju trombociti koji stvaraju trombocitne čepove u mikrovaskulaturi, a to dovodi i do oštećenja eritrocita pri njihovom prolasku. Zbog toga su dva temeljna obilježja TTP-a potrošna trombocitopenija i hemolitička anemija. Promjene su najizraženije u mikrovaskulaturi mozga i bubrega gdje trombocitni čepovi dovode do okluzije i posljedične ishemije, zbog čega TTP najčešće prate neurološke manifestacije i akutno bubrežno oštećenje.
Klinička slika. Klasično očitovanje TTP-a obuhvaća pentadu simptoma koja uključuje trombocitopeniju, mikroangiopatsku hemolitičku anemiju, febrilno stanje, neurološke manifestacije te akutno bubrežno oštećenje, iako je ono prisutno kod manje od jedne trećine bolesnika, a najčešće se dijagnosticira na osnovi laboratorijskih nalaza koji uključuju trombocitopeniju i hemolitičku anemiju. Bolesnici razvijaju dvije skupine kliničkih manifestacija: jednu vezanu uz tromboze i ishemijske incidente posljedično formiranju trombocitnih čepova, te drugu vezanu hemoragijsku dijatezu koja proizlazi iz potrošne koagulopatije. Ishemiji su najskloniji središnji živčani sustav (neurološke manifestacije) te bubrezi (akutno bubrežno oštećenje), dok se hemoragijska dijateza najčešće očituje krvarenjima u koži i potkoži (tzv. „suha krvarenja“) u obliku petehija, ekhimoza i sufuzija, dok su velika krvarenja (tzv. „mokra krvarenja“) rjeđa pojava.
Dijagnoza se prvenstveno postavlja na osnovi kliničke slike i laboratorijskih nalaza. U laboratorijskim nalazima bolesnika prisutna je umjereno izražena trombocitopenija (vrijednosti trombocita obično se kreću između 10 i 30 x 109/L) te hemolitička anemija karakterizirana sniženim vrijednostima hemoglobina i haptoglobina, retikulocitoza te povećane vrijednosti laktat-dehidrogenaze i bilirubina. U razmazu periferne krvi mogu se naći shizociti (Slika 6.4.). Standardni koagulogram (PV, aPTV, fibrinogen, antitrombin III) bolesnika je uredan. Posljedično akutnom bubrežnom oštećenju dolazi do porasta ureje i kreatinina, a ovisno o njegovoj težini javljaju se i metabolička acidoza i/ili hiperkalijemija. Promjene na središnjem živčanom sustavu potvrđuju se nalazom CT-a ili MR-a mozga. Konačna potvrda bolesti zasniva se na dokazu nedostatka ili smanjene aktivnosti ADAMTS13 proteina (< 5 %) i/ili potvrdom prisustva inhibitora ili antitijela IgG klase na ADAMTS13 (navedeno testiranje ne radi se rutinski u svim laboratorijima).
Slika 6.4. Nalaz shizocita u razmazu periferne krvi
Liječenje. Osnovu liječenja čini terapijska plazmafereza koja omogućava uklanjanje antitijela na ADAMTS13, kao i kompleksa antitijelo-ADAMTS13, a samom nadoknadom plazme vrši se i istodobna suplementacija ADAMTS13 metaloproteinaza. Taj je postupak smanjio stopu smrtnosti s 90 na 10 – 15 %. Postupak je potrebno započeti što ranije, obično po shemi izmjene 1,5 volumena plazme kroz tri dana, a potom se nastavlja s izmjenom jednostrukoga volumena plazme do potpune remisije bolesti, odnosno oporavka broja trombocita za što je potrebno i do deset do četrnaest dana (nakon postizanja remisije preporuča se još dva dana provođenje plazmafereze). Osim plazmafereze, indicirana je i intravenska primjena kortikosteroida (metilprednizolon), a u rezistentnim slučajevima indicirano je i liječenje rituksimabom (monoklonalno antitijelo usmjereno na CD20), a moguća je i primjena kaplacizumaba koji inhibira interakciju između ultravelikih multimera von Willenbrandova faktora i trombocita. Primjena transfuzije trombocita kod ovih je bolesnika kontraindicirana jer povećava rizik od trombotskih incidenata i ishemijskih oštećenja. Liječenje kongenitalnog oblika TTP-a provodi se supstitucijom ADAMTS13.
HEMOLITIČKO-UREMIČNI SINDROM
Definicija. Hemolitičko-uremični sindrom (HUS) također predstavlja oblik potrošne trombocitopenije koja se prezentira zajedno s hemolitičkom anemijom te ga je ponekad teško diferencirati od trombotičke trombocitopenične purpure. Za razliku od TTP-a, zahvaćenost mozga je izuzetno rijeka, dominira akutno bubrežno oštećenje, a intravaskularna tromboza potaknuta je raznim bakterijskim toksinima ili lijekovima uz očuvanu aktivnost ADMTS13 metaloproteinaza.
Etiopatogeneza. HUS se razvija kao posljedica direktnoga oštećenja endotelnih stanica koje može biti posredovano bakterijskim toksinima ili lijekovima s posljedičnim pojačanim otpuštanjem von Willenbrandova faktora koji potiče aktivaciju trombocita, stvaranje trombocitnih čepova i hemolitičke anemije povezane s njima, potrošne trombocitopenije i protrombotske dijateze (ishemija izazvana trombocitnim čepovima). Klasično razlikujemo dva tipa ovog sindroma: tipični i atipični HUS. Tipični HUS (90 %) obično nastaje kod djece nakon gastrointestinalne infekcije (E. colli, Shigella dysenteriae) potaknut bakterijskim toksinima, dok atipični HUS (10 %) obično pogađa odrasle, a najčešće se razvija nakon respiratornih infekcija (S. pneumoniae) te nakon uzimanja nekih lijekova (oralni kontraceptivi, ciklosporin, cisplatina).
Klinička slika i dijagnoza. Kliničkom slikom bolesnika dominira akutna bubrežna ozljeda uz trombocitopeniju i hemolitičku anemiju, a ovisno o zahvaćenosti, prisutni su i simptomi i znakovi drugih organa (neurološke manifestacije kao posljedice trombotskih incidenata rijetke su kod ovih bolesnika). Dijagnoza se postavlja na temelju anamnestičkih podataka te kliničkih i laboratorijskih nalaza koji uključuju trombocitopeniju, hemolitičku anemiju te poremećaj bubrežne funkcije. Glavno diferencijalno dijagnostičko razgraničenje TTP-a i HUS-a omogućeno je određivanjem koncentracije i/ili aktivnosti ADAMTS13 ili određivanjem prisutnosti specifičnih protutijela na ADAMTS13. Ovisno o kliničkim manifestacijama, treba provesti i drugu specifičnu dijagnostiku (npr. CT ili MR mozga i sl.).
Liječenje bolesnika uključuje primjenu odgovarajuće antimikrobne terapije te primjenu postupka plazmafereze, iako je njezin terapijski učinak znatno slabiji nego kod bolesnika s TTP-om. Kod bolesnika koji ne reagiraju na te terapijske metode u obzir dolazi i primjena eculizumaba.
LIJEKOVIMA INDUCIRANA TROMBOCITOPENIJA
Definicija i etiopatogeneza. Lijekovima inducirana trombocitopenija jedan je od najčešćih oblika izoliranih trombocitopenija u kliničkoj praksi. Temeljni patogenetski mehanizam razvoja te trombocitopenije imunološke je prirode, a podrazumijeva vezanje lijeka ili njegova metabolita na površinu trombocita pri čemu nastaje neoantigen kojega imunološki sustav prepoznaje i pokreće reakciju protiv njega. Drugi, rjeđi mehanizmi nastanka trombocitopenije inducirane lijekovima podrazumijevaju učinak lijekova na samu koštanu srž smanjivanjem proizvodnje trombocita (citostatici) ili nekim drugim specifičnim mehanizmima, kao što je to slučaj pri trombocitopeniji induciranoj heparinom (kasnije opisana). Različiti lijekovi mogu biti povezani s nastankom trombocitopenije: 1) antitrombocitni lijekovi (abciksimab, anagrelid, tiklopidin, eptifibatid); 2) antimikrobni lijekovi (adefovir, indinavir, ritonavir, flukonazol, izoniazid, linezolid, penicilini, sulfonamidi, rimfampicin, vankomicin); 3) kardiovaskularni lijekovi (amiodaron, atorvastatin, simvastatin, kaptopril, digoksin, hidroklortiazid), 4) analgetici (acetaminofen, diklofenak, naproksen); 5) gastrointestinalni lijekovi (cimetidin, ranitidin); 6) neuropsihijatrijski lijekovi (karbamazepin, haloperidol, fenitoin); 7) antikoagulantni lijekovi (klasični i niskomolekularni hearini) te 8) imunosupresivi, imunomodulatori i kemoterapeutici.
Klinička slika i dijagnoza. Tipično kliničko očitovanje trombocitopenije inducirane lijekovima podrazumijeva razvoj umjerene do teške trombocitopenije praćene pojavom mukokutanoga krvarenja (hematomi, sufuzije, ekhimoze) u vremenskom periodu od sedam do četrnaest dana od početka izlaganja lijeku. Prestankom primjene lijeka oporavak broja trombocita se u većini slučajeva bilježi kroz sedam do deset dana, iako ono može biti i dulje (u ovisnosti o bubrežnoj i jetrenoj funkciji u ogledu metaboliziranja i eliminacije lijeka). Dijagnoza se postavlja na osnovi kliničke slike i anamnestičkih podataka o povezanosti trombocitopenije s uzimanjem (ili prekidom uzimanja) lijeka. Nema specifičnih laboratorijskih testova kojima bi se potvrdila dijagnoza ove bolesti.
Liječenje se kod većine bolesnika sastoji od prekidanja uzimanja lijeka za koji se smatra da je povezan s razvojem trombocitopenije. Kod bolesnika s teškom trombocitopenijom i/ili manifestnim krvarenjem indicirana je primjena transfuzije trombocita. Ukoliko se ne može sa sigurnošću isključiti da se radi o idiopatskoj (imunoj) trombocitopeniji, treba primijeniti odgovarajuće terapijske mjere kako je to prethodno opisano.
HEPARINOM INDUCIRANA TROMBOCITOPENIJA
Definicija i epidemiologija. Heparinom inducirana trombocitopenija (HIT) poseban je oblik lijekovima inducirane trombocitopenije kojega se povezuje s primjenom heparina, a može biti udružen s pojavom arterijskih i venskih trombotskih incidenata. HIT se javlja kod oko 2 do 5 % bolesnika na terapiji klasičnim (nefrakcioniranim) heparinom te kod 0,5 od 0,7 % bolesnika na terapiji niskomolekularnim heparinima. Razvoj HIT-a gotovo se nikada ne susreće kod bolesnika koji su na terapiji fondaparinuksom.
Etiopatogeneza. Mehanizam razvoja HIT-a uključuje složenu imunološki posredovanu reakciju koja se sastoji od nekoliko koraka. Primarni događaj u ovoj reakciji uključuje vezanje trombocitnoga faktora 4 (PF4, engl. platelet factor 4) kojega luče trombociti za molekulu heparina, pri čemu nastaje heparin-PF4 kompleks na kojemu se stvaraju specifična IgG antitijela i vežu se za njih. Novonastali kompleks kojega čini specifično IgG antitijelo i ranije opisan heparin-PF4 kompleks veže se za Fc receptore na površini trombocita te ih aktivira. Aktivacija trombocita dovodi do njihove agregacije i stvaranja trombocitnih čepova koji su odgovorni za ishemijske manifestacije, a posljedično trošenju trombocita razvija se potrošna trombocitopenija koja se povezuje s hemoragijskom dijatezom. Za razliku od ranije opisanoga imunološki posredovanog mehanizma, HIT može nastati i neimunološki posredovanom reakcijom, odnosno izravnom interakcijom heparina s površinom trombocita što dovodi do njihove aktivacije. Osnovne razlike između ta dva tipa prikazane su u Tablici 6.14.
|
Tablica 6.14. Karakteristike HIT-a
|
|
Karakteristike
|
Neimimunološki posredovani HIT
(HIT tip I)
|
Imunološki posredovan HIT (HIT tip II)
|
|
Uzrok
|
Direktni kontakt s heparinom
|
Prisutnost autoantitijela
|
|
Incidencija
|
5 do 30 %
|
1 do 3 %
|
|
Vremensko pojavljivanje
|
Unutar 4 dana
|
5 do 10 dana
|
|
Komplikacije
|
Nema
|
Trombotski incidenti
|
|
Oporavak
|
1 do 3 dana
|
5 do 7 dana
|
Klinička slika i dijagnoza. Na HIT treba pomišljati kod bolesnika kod kojih se trombocitopenija razvije tijekom liječenja heparinom, osobito klasičnim (nefrakcioniranim) heparinom. Ona se razvija unutar prva dva tjedna liječenja (obično unutar pet do deset dana, iako može i ranije kod bolesnika koji su prethodno senzibilizirani). S obzirom na širok raspon referentnih vrijednosti trombocita (150 – 450 x 109/L), na HIT treba pomišljati kod bolesnika koji razviju 50 %-tno smanjenje broja trombocita u odnosu na početne vrijednosti tijekom liječenja heparinom, bez obzira na to što oni još uvijek mogu biti unutar referentnih vrijednosti. Klinički, bolest je obično asimptomatska, a mogu se javiti ishemijske manifestacije (neurološke, bubrežne) te znakovi hemoragijske dijateze. Osim kliničke slike, laboratorijskoga nalaza smanjenoga broja trombocita i anamnestičke sumnje na HIT, on se potvrđuje prisutnošću tzv. HIT protutijela (IgA protutijela na kompleks heparin-PF4), iako on može biti prisutan i kod bolesnika koji primaju heparin, a nemaju klinički manifestnu bolest.
Liječenje podrazumijeva obustavu primjene heparina te sprečavanje nastanka trombotskih komplikacija povezanih s HIT-om. Nastalo prokoagulantno stanje zbrinjava se primjenom direktnih inhibitora trombina (argatroban, bivalirudin) ili fondaprinuksa (iako to nije službeno potvrđeno), sve dok se broj trombocita ne oporavi, a potom se nastavlja liječenje varfarinom uz održavanje ciljnih vrijednosti INR-a između 2 i 3 kroz sljedećih 30 dana, odnosno tri do šest mjeseci ukoliko bolesnici imaju povećan rizik. Kod bolesnika je potrebno izbjegavati daljnju primjenu heparina, a ukoliko je ona neophodna, opravdana je samo kod bolesnika kojima se ELISA testom potvrdi odsustvo HIT antitijela.
OSTALI OBLICI TROMBOCITOPENIJA
U kliničkoj su praksi česti i drugi oblici trombocitopenija, kao što su diseminirana intravskularna koagulopatija te trombocitopenija u sepsi, što je opisano u poglavlju „Anemija, trombocitopenija i koagulopatija kod kritično oboljelih bolesnika“.
TROMBOCITOZA
Definicija. Trombocitoza je naziv za stanje povećanoga broja trombocita i to u vrijednosti većoj od 450 x 109/L, a prema etiologiji se razlikuju tri oblika trombocitoze: fiziološka, reaktivna i klonalna. Pseudotrombocitoza ili lažna trombocitoza rijetko se može vidjeti, i to kod bolesnika s krioglobulinemijom kada brojač istaložene cirkulirajuće proteine broji kao trombocite. Kod svih bolesnika s nalazom trombocitoze treba ponoviti uzorkovanje krvi kako bi se utvrdilo radi li se o prolaznom ili perzistentnom poremećaju.
Fiziološka trombocitoza označava umjereno, prolazno povećanje broja trombocita koje možemo vidjeti u stanjima povećanoga fizičkog napora, stresa i drugih stanja praćenih pojačanom aktivnosti simpatikusa.
Reaktivna (sekundarna) trombocitoza može se vidjeti pri različitim bolestima i stanjima: akutno krvarenje, hemolitička i sideropenična anemija, akutne infekcije, kronične upalne bolesti, maligne bolesti, postoperativna i posttraumatska stanja te primjena određenih lijekova (vinkristin, faktori rasta). Reaktivna trombocitoza obično je prolazne prirode, pojavljuje se uz stanja za koja se zna da mogu biti povezana s povećanjem broja trombocita te bolesnici nemaju izraženih znakova mijeloproliferativnoga sindroma niti splenomegaliju.
Klonalna (primarna) trombocitoza pojavljuje se u sklopu opisanih mijeloproliferativnih oboljenja, kao što su esencijalna trombocitoza, policitemija rubra vera, kronična mijeloična leukemija, akutna mijeloična leukemija te idiopatska mijelofibroza. Osnovne značajke klonalne trombocitoze su perzistencija patoloških nalaza, prisutnost simptoma i znakova koji ukazuju na mijeloproliferativnu bolest te prisutna splenomegalija.
POREMEĆAJI FUNKCIJE TROMBOCITA
Kongenitalni poremećaji funkcije trombocita rijetko se susreću u kliničkoj praksi, posebice u odrasloj populaciji, s obzirom na to da se oni manifestiraju hemoragijskom dijatezom u mlađoj životnoj dobi, kada ih se i dijagnosticira. Dva najčešća oblika kongenitalnoga poremećaja funkcije trombocita su Bernard-Soulierov sindrom te Glanzmannova trombastenija. Bernard-Soulierov sindrom predstavlja autosomno recesivni nasljedni poremećaj ekspresije glikoproteinskoga receptora Ib/IX (receptor za von Willenbrandov faktor), zbog čega bolesnici pate od poremećaja aktivacije trombocita, dok kod Glanzmannove trombastenije nalazimo poremećaj IIb/IIIa receptora koji ima ključnu ulogu u procesu agregacije i međusobnoga povezivanja trombocita. Posljedično poremećaju aktivacije i/ili agregacije trombocita bolesnici razvijaju sklonost krvarenju, a u laboratorijskim nalazima obično je prisutno samo produljeno vrijeme krvarenja uz uredne ostale koagulacijske parametre. Na poremećaj funkcije trombocita ukazuju i patološki nalazi testova agregacije trombocita. S obzirom na to da je većina ovih bolesnika asimptomatska, nije potrebno specifično liječenje. Kod krvarenja se bolesnicima daju transfuzije trombocita, a pozitivan učinak pokazuju i primjena dezmopresina, rekombinantnoga humanog aktivatora faktora VII te antifibrinolitičkih lijekova.
Stečeni poremećaji funkcije trombocita susreću se češće od kongenitalnih, a najčešće se pojavljuju kod odraslih osoba i povezani su s primjenom antitrombocitnih lijekova. Ako je funkcija trombocita ireverzibilno inhibirana, oporavak funkcije trombocita očekuje se kroz osam do deset dana nakon prestanka primjene lijeka. Kod reverzibilne inhibicije trombocita, oporavak funkcije trombocita zbiva se po eliminaciji lijeka iz organizma. Specifično liječenje nije potrebno, osim kod bolesnika s krvarenjem, a sastoji se od transfuzije trombocita.
BENIGNE BOLESTI I POREMEĆAJI LEUKOCITA
Leukociti ili bijele krvne stanice nositelji su imunološke obrane organizma. Njihove normalne vrijednosti u perifernoj krvi kreću se između 4,5 i 10 x 109/L sa srednjom vrijednosti od oko 7,5 x 109/L. Njihov najveći dio čine neutrofili (oko 60 %), a uz njih nalazimo i limfocite, monocite, bazofile i eozinofile. Poremećaje broja leukocita nazivamo leukocitozom (povećan broj) i leukopenijom (smanjeni broj), a mogu se javiti kao primarni poremećaj (hematološko oboljenje) ili sekundarni (posljedično drugom oboljenju). Najznačajniji poremećaji iz ove skupine u kliničkoj praksi su neutrofilija, neutropenija te eozinofilija.
NEUTROFILIJA
Definicija. Neutrofilija je najčešća vrsta leukocitoze, a o njoj govorimo kada apsolutni broj neutrofila prelazi 7,5 x 109/L. Apsolutni broj neutrofila označava se kao umnožak ukupnoga broja leukocita i postotka neutrofila u diferencijalnoj krvnoj slici.
Etiologija. Neutrofilija može biti posljedica različitih bolesti i stanja kao što su: 1) infekcije (bakterijske, virusne, gljivične), 2) bolesti vezivnoga tkiva (vaskulitis, reumatoidni artritis) i kronične upalne bolesti, 3) zloćudne bolesti (solidni maligni tumori, Hodgkinova bolest i ne-Hodgkinovi limfomi), 4) hematološki poremećaji i oboljenja (mijelodisplastični sindrom, akutne i kronične mijeloproliferativne bolesti), 5) lijekovi (kortikosteroidi, adrenalin, litij, faktori rasta, klozapin, dezmopresin), 6) kemijski spojevi (trovanje živom, olovom ili etilen-glikolom), 7) metabolički poremećaji (laktacidoza, dijabetička ketoacidoza, tireotoksikoza, Cushingov sindrom, uremija, giht), 8) pušenje, 9) trauma i tkivna nekroza te 10) ostali rijetki oblici (hereditarna neutrofilija, kronična idiopatska neutrofilija). Neki od najznačajnijih uzroka i posebnih oblika neutrofilija opisani su posebno.
Infekcije. Bakterijske su infekcije najčešći uzroci leukocitoze s neutrofilijom. Broj leukocita kod takvih bolesnika obično se kreće između 10 i 25 x 109/L, iako neki uzročnici (inkapsulirani sojevi) mogu dovesti i do znatno izraženije leukocitoze. Karakteristično je da takvi bolesnici imaju povećan broj (udio) nesegmentiranih neutrofila što se naziva skretanjem ulijevo. U fiziološkim okolnostima u krv se iz koštane srži otpuštaju segmentirani neutrofili, dok se u stanjima povećane potrebe za njima oni otpuštaju nepotpuno zreli, odnosno nesegmentirani. Ipak, skretanje ulijevo nije patognomonični znak za bakterijske infekcije, nego ga se može naći i u nekim drugim bolestima i stanjima (oštećenje i nekroza tkiva, mijeloproliferativne bolesti, metabolički poremećaji, reakcije preosjetljivosti i sl.). Treba napomenuti i da ne moraju nužno sve bakterijske infekcije biti praćene leukocitozom i neutrofilijom, a smanjeni odgovor mogu imati i starije osobe i kronični bolesnici. Virusne infekcije također mogu biti praćene neutrofilijom, ali je kod njih ukupni broj leukocita najčešće uredan ili snižen.
Lijekovi. Primjena određenih lijekova može biti povezana s razvojem neutrofilije i uz bakterijsku se infekciju smatra jednim od najčešćih uzroka. Kortikosteroidi su najčešće primjenjivani lijekovi koji mogu dovesti do leukocitoze i neutrofilije. Ona je obično prolazna, a broj leukocita ne prelazi 20 x 109/L. Porast broja leukocita može se zamijetiti već nekoliko sati od primjene terapije, osobito ako se radi o parenteralnom unosu lijeka. Za razliku od neutrofilije u bakterijskim infekcijama, ovi su neutrofili zreli, odnosno segmentirani (bez fenomena skretanja ulijevo).
Pseudoneutrofilija. S obzirom na to da se zreli neutrofili osim u koštanoj srži i perifernoj krvi nalaze i u rezervnom obliku, adherirani za male krvne žile u različitim stanjima, oni se mogu otpustiti u perifernu krv što će povisiti njihovu vrijednost u krvi, ali ne i njihovu ukupnu količinu u tijelu, pa se taj oblik neutrofilije naziva pseudoneutrofilijom, ili lažnom ili fiziološkom neutrofilijom. Ona može biti prisutna u stanjima tjelesnoga opterećenja, stresa, traume, hipoksije i prekomjernoga pušenja.
Leukemoidna reakcija naziv je za reaktivnu neutrofiliju koja predstavlja odgovor normalne koštane srži na neki infektivni ili neinfektivni podražaj, a karakterizirana je pojavom izražene leukocitoze (> 25 – 30 x 109/L) i neutrofilije, što može nalikovati na neoplastičnu neutrofiliju. To se stanje može razviti posljedično infekciji, traumi ili krvarenju. Iako broj leukocita ispod 50 x 109/L, kao i njegovo vidljivo smanjenje nakon započetoga liječenja (npr. antimikrobne terapije), sugerira da se radi o leukemoidnoj reakciji, a ne o mijeloproliferativnoj bolesti, glavnu diferencijalno dijagnostičku ulogu imaju biopsija i analiza koštane srži koja je uredna (ukazuje na granulocitnu hiperplaziju) kod bolesnika s leukemoidnom reakcijom.
Hereditarna neutrofilija označava rijetku autosomno-dominantnu nasljednu bolest obilježenu pojačanim stvaranjem neutrofila. Nastaje uslijed mutacije GCF3 gena što za posljedicu ima prekomjernu aktivaciju G-CSF receptora, a to u konačnici rezultira pojačanim stvaranjem neutrofila. Stvoreni neutrofili imaju urednu funkciju te takvi bolesnici nemaju sklonosti razvoju infekcija ili drugih posljedica.
Kronična idiopatska neutrofilija označava kliničko stanje obilježeno izraženo neutrofilijom periferne krvi u rasponu od 11 do 40 x 109/L, uz uredan nalaz biopsije koštane srži. Dosadašnja praćenja takvih bolesnika nisu pokazala da to stanje dovodi do značajnijih kliničkih posljedica.
Klinički pristup bolesniku s neutrofilijom. U pristupu bolesniku s leukocitozom i neutrofilijom osnovno je razgraničiti radi li se o mijeloproliferativnom poremećaju ili sekundarnoj neutrofiliji. U tome pomaže razmaz periferne krvi: ukoliko se u njemu otkrije leukoeritroblastoza (pojava nezrelih prekursora eritrocita i leukocita), postavlja se sumnja na mijeloproliferativni poremećaj te je indicirana biopsija koštane srži. Kod bolesnika s urednim nalazom razmaza periferne krvi najčešći uzrok neutrofilije su infekcije na što upućuju vrućica i odgovarajuća klinička slika. Ukoliko neutrofiliju s urednim razmazom periferne krvi ne prati klinička slika infekcije, treba pomišljati na ostale sekundarne uzroke neutrofilije (primjena lijekova, autoimune bolesti, kronične upalne bolesti).
NEUTROPENIJA
Definicija. Neutropenija je uz limfocitopeniju najčešći uzrok leukopenije, odnosno ukupnoga smanjenja broja leukocita, a definira se kao smanjenje apsolutnoga broja neutrofila ispod 1,5 x 109/L, a apsolutni broj neutrofila označava se, kako je to već navedeno, kao umnožak ukupnoga broja leukocita i postotka neutrofila u diferencijalnoj krvnoj slici. Pri tumačenju nalaza apsolutnoga broja neutrofila treba imati na umu da osobe određenih etničkih pripadnosti fiziološki mogu imati smanjene vrijednosti broja neutrofila (25 % odraslih, zdravih crnaca ima apsolutni broj neutrofila između 1,0 i 1,5 x 109/L).
Klasifikacija. Prema vrijednostima apsolutnoga broja neutrofila, neutropeniju dijelimo u tri stupnja: 1) blaga neutropenija (1,0 - 1,5 x 109/L), umjerena neutropenija (0,5 - 1,0 x 109/L) te teška neutropenija (< 0,5 x 109/L). Učestalost i rizik od nastanka infekcije uvjetovane neutropenijom obrnuto su proporcionalni vrijednosti njihovoga apsolutnog broja tako da kod vrijednosti neutrofila ispod 1,0 x 109/L učestalost infekcija, posebice bakterijskih, značajno raste.
Etiologija. Neutropenija može biti uzrokovana različitim bolestima i stanjima: 1) infekcije, 2) lijekovi, 3) hematološka oboljenja i potiskivanje koštane srži, 4) splenomegalija i hipersplenizam, 5) nutritivni deficiti te 6) specifični oblici (imuna neutropenija, kronična idiopatska neutropenija, teška kongenitalna neutropenija, ciklička neutropenija).
Infekcije su pored lijekova jedan od najčešćih uzroka nastanka neutropenije. Ona je obično povezana s virusnim infekcijama koje uzrokuju prolaznu neutropeniju (vodene kozice, ospice, hepatitis A i B, gripa, infekcije citomegalovirusom i Epstein-Barrovim virusom). Neutropenija može biti prisutna i uslijed bakterijskih infekcija, ali znatno rjeđe (obično je povezana sa septičnim stanjem i predstavlja loš prognostički znak).
Lijekovi su također među češćim uzrocima neutropenije, a može se raditi o predvidivoj ili nepredvidivoj (idiosinkratičnoj) reakciji. Predvidiva neutropenija obično se viđa kod bolesnika koji primaju citotoksične kemoterapeutike. Lijekovima izazvana neutropenija obično se javlja kroz nekoliko dana do nekoliko tjedana od početka primjene, a može se javiti u sklopu dugotrajnoga uzimanja lijekova. Mehanizam nastanka može biti višestruk: smanjeno stvaranje (mijelosupresija), pojačana destrukcija ili ubrzano uklanjanje. Oporavak broja neutrofila očekuje se kod gotovo svih bolesnika, a zbiva se tjedan do dva od prestanka primjene lijeka. Lijekovi koji se najčešće povezuju s neutropenijom su citotoksični kemoterapeutici, antibiotici (penicilini, sulfonamidi, cefalosporini, kloramfenikol), nesteroidni protuupalni lijekovi, antitireoidini lijekovi (propiltiouracil, metimazol), alfa-metildopa, alopurinol, cimetidin, klozapin i neki diuretici (spirinolakton, klorotiazid).
Hematološke bolesti i potiskivanje koštane srži. Različita hematološka oboljenja mogu biti uzrok neutropenije, na primjer akutna ili kronična leukemija, aplastična anemija, sindrom mijelodisplazije, kao i patološki procesi koji dovode do potiskivanja normalnoga hematopoetskog tkiva (tumori, granulomatoza, mijelofibroza).
Imuna neutropenija nastaje kao posljedica izravne destrukcije neutrofila posredovane specifičnim antineutrofilnim antitijelima, što se može javiti kao primarni poremećaj (obično kod djece) ili sekundarno kod bolesnika s drugim autoimunim oboljenjima (hipertireoza, Wegenerova granulomatoza, reumatoidni artritis, sistemski eritematozni lupus).
Kronična idiopatska neutropenija definira se kao stečeni poremećaj granulocitopoeze obilježen produljenim periodom smanjenoga stvaranja neutrofila uz odsutnost drugih mogućih uzroka. Neutropenija je kod takvih bolesnika obično blaga i nema većega odraza na kliničko stanje bolesnika. Iako je točan uzrok bolesti nepoznat, kod većine bolesnika mogu se naći određeni polimorfizmi TGF-B gena. Bolesnici u većini slučajeva imaju povoljan klinički tijek, a samo manji broj će zahtijevati primjenu faktora rasta granulocita.
Teška kongenitalna neutropenija naziv je za heterogenu skupinu genetskih poremećaja koji rezultiraju zastojem u sazrijevanju neutrofila i posljedičnom neutropenijom koja se javlja već u prvim mjesecima života i praćena je učestalim, teškim bakterijskim infekcijama. Najčešće se radi o mutacijama ELANE, HAX1, WASP i TAZ gena.
Ciklična neutropenija podrazumijeva rijetko kliničko stanje obilježeno povremenim epizodama neutropenije koje traju tri do pet dana, a javljaju se u vremenskim intervalima od otprilike tri tjedna. Za vrijeme neutropenije bolesnici obično imaju vrućicu i aftozne promjene usne šupljine (rijetko i kože te sluznice dišnoga sustava). Većinom se radi o nasljednom poremećaju uvjetovanom mutacijom gena za neutrofilnu esterazu (ELANE gen). Dijagnoza se postavlja na osnovi kliničke slike i opetovanoga određivanja broja neutrofila. Kod takvih je bolesnika u odrasloj dobi nešto viša učestalost razvoja T-staničnih limfoma.
Ostali uzroci. Splenomegalija i hipersplenizam često su praćeni blagom do umjerenom neutropenijom uz koju obično nalazimo i anemiju i trombocitopeniju. Nedostatak folata i vitamina B12 također može biti uzrok nastanka neutropenije koja se tada prezentira zajedno s megaloblastičnom anemijom.
Klinički pristup bolesniku s neutropenijom. Bolesnici s blagom neutropenijom obično nemaju simptoma, dok se kod onih s umjerenom i teškom češće javljaju infekcije, i to najčešće dišnoga i mokraćnog sustava s tendencijom brze progresije u sepsu i septički šok. Poseban klinički entitet čini febrilna neutropenija opisana u poglavlju „Hitna stanja u hematologiji“. Neutropenija najčešće nastaje kao posljedica infekcije (virusne ili teške bakterijske) te primjene lijekova, što se mora prvo uzeti u obzir kod bolesnika kada ju se otkrije u laboratorijskim nalazima. Ukoliko bolesnik ima neutropeniju i znakove infekcije, indicirana je što ranija primjena antibiotika, a ako je broj neutrofila manji od 0,5 x 109/L, indicirana je i primjena faktora rasta granulocita (G-CSF, filgrastim). Sve lijekove koji se povezuju s neutropenijom treba izostaviti iz terapije i zamijeniti ih drugim odgovarajućim lijekom. Ukoliko bolesnik ima neutropeniju bez znakova infekcije te ne uzima lijekove koji se povezuju s njom, indicirana je šira dijagnostička obrada koja uključuje biopsiju/punkciju koštane srži, određivanje vrijednosti folata i vitamina B12 te probir na autoimune bolesti. Kod bolesnika s obiteljskom anamnezom pojavnosti neutropenije u obzir dolaze i drugi, rjeđi oblici neutropenije, kao što su ciklička neutropenija ili kronična idiopatska neutropenija. Liječenje granulocitnim faktorima rasta indicirano je kod simptomatskih bolesnika (učestale infekcije).
EOZINOFILIJA I HIPEREOZINOFILNI SINDROMI
Definicija. Eozinofilija se definira apsolutnim brojem eozinofila većim od 0,5 x 109/L, a apsolutni broj eozinofila označava umnožak ukupnoga broja leukocita i postotka eozinofila u diferencijalnoj krvnoj slici. Eozinofilni sindromi predstavljaju heterogenu skupinu bolesti povezanih s eozinofilijom, a očituju se eozinofilnom infiltracijom različitih tkiva i organa.
Etiologija. Uzroci eozinofilije mogu biti razni, a može ih se svrstati u tri skupine: reaktivna eozinofilija, eozinofilija udružena s drugim bolestima te primarna eozinofilija.
Reaktivna eozinofilija najčešći je oblik eozinofilije, a javlja se uslijed specifičnih podražaja. Najčešći oblici te vrste eozinofilije su: 1) alergijske bolesti kao što su astma, atopijski dermatitis, alergijski rinitis, 2) reakcije preosjetljivosti na lijekove te 3) infekcije parazitima, rjeđe virusima (virus humane imunodeficijencije) ili gljivicama (alergijska bronhopulmonalna aspergiloza, kokcidiomikoza).
Eozinofilija udružena s drugim bolestima. Eozinofiliju možemo naći u različitim bolestima i stanjima kao što su: 1) gastrointestinalne eozinofilije (eozinofilni ezofagitis, eozinofilni gastroenteritis), 2) kožne bolesti (bulozni pemfigoid, urtikarija, eozinofilni celulitis, eozinofilni angioedem), 3) plućne bolesti (eozinofilna pneumonija, alergijska bronhopulmonalna aspergiloza), 4) bolesti središnjega živčanog sustava (eozinofilni meningitis), 5) autoimune bolesti (Churg-Straussov sindrom, eozinofilni fasciitis), 6) maligne bolesti (solidni tumori, Hodgkinova bolest), 7) stanje nakon transplantacije te reakcije odbacivanja transplantata.
Primarna eozinofilija označava eozinofiliju koja nastaje kao posljedica klonalne proliferacije eozinofila.
Idiopatski hipereozinofilni sindrom definira se kao dugotrajna hipereozinofilija (> 1,5 x 109/L tijekom šest mjeseci) uz simptome i znakove zahvaćanja drugih organa, u odsutnosti drugih poznatih uzroka eozinofilije. Danas se zna da se kod velikoga broja takvih bolesnika mogu naći specifične genetske promjene od kojih je najznačajnija pojava FIP1L1-PDGFRA fuzijskoga gena udružena s mikrodelecijom 4q24, te njegovo prisustvo u uzroku koštane srži danas držimo kriterijem za postavljanje dijagnoze neoplastične varijante hipereozinofilnog sindroma (HES), mijeloidna varijanta.
Klinički pristup bolesniku s eozinofilijom. Kod bolesnika s eozinofilijom najprije je potrebno isključiti reaktivni oblik koji je najčešće povezan s atopijskim oboljenjima, primjenom lijekova te parazitarnim infestacijama. Kada je isključena reaktivna eozinfolija, potrebno je provesti dijagnostičku obradu u smislu utvrđivanja postojanja druge eozinofilne infiltracije (ehokardiografija, CT prsnog koša i trbuha, endoskopske pretrage, biopsija i sl.), kao i primarne eozinofilije (biopsija koštane srži). Osnovna pretraga pri biopsiji koštane srži je utvrđivanje postojanosti FIP1L1-PDGFRA fuzijskoga gena te klonalnosti eozinofila. Ukoliko je FIP1L1-PDGFRA fuzijski gen pozitivan ili je prisutna klonalnost, govorimo o mijeloidnoj varijanti HES-a, dok kod bolesnika s negativnim FIP1L1-PDGFRA fuzijskim genom i bez klonalnosti govorimo o idiopatskom hiperozinofilnom sindromu. Prisutnost FIP1L1-PDGFRA fuzijskoga gena ima i terapijsko značenje, s obzirom na to da je liječenje izbora za takve bolesnike primjena imatiniba, a kod bolesnika koji ne reagiraju na njega dobre rezultate pokazuje i sorafenib. Primjenom tih lijekova smanjenje broja eozinofila moguće je već unutar prvoga tjedna liječenja. Kod bolesnika s limfocitnom varijantom HES-a i idiopatskim HES-om liječenje se temelji na primjeni kortikosteroida. Kod pacijenata kod kojih se uz prethodno navedenu terapiju ne postiže optimalni odgovor u fazi je istraživanja primjena monoklonalnih anti-IL-5 antitijela (mepolizumab, reslizumab) koja inhibiraju diferencijaciju eozinofila te njihovo oslobađanje iz koštane srži što je posredovano interleukinom-5 (IL-5).
OSTALI POREMEĆAJI BROJA LEUKOCITA
Limfocitoza je naziv za povećanje apsolutnoga broja limfocita iznad 4 x 109/L, a apsolutni broj limfocita definira se kao umnožak ukupnoga broja leukocita i postotka limfocita u diferencijalnoj krvnoj slici. Najčešći uzrok limfocitoze je infekcija, a najčešće se radi o virusnim infekcijama (Epstein-Barrov virus, citomegalovirus, adenovirusi, virusi hepatitisa, virus humane imunodeficijencije) te nekim specifičnim bakterijskim infekcijama (pertusis, bruceloza, tifus, tuberkuloza). Ostali uzroci limfocitoze uključuju reakciju preosjetljivosti na lijekove, serumsku bolest, maligna oboljenja (akutna i kronična limfocitna leukemija, limfomi), prekomjeran fizički napor, endokrine bolesti (hipopituitarizam, insuficijencija nadbubrežne žlijezde) te prolazna stresna stanja (trauma, infarkt miokarda, srčani zastoj, porod, operacije i sl.).
Monocitoza je naziv za stanje obilježeno povećanim apsolutnim brojem monocita iznad 0,75 x 109/L, što može biti uzrokovano hematološkim poremećajima (akutna i kronična mijeloična leukemija, mijelodisplastični sindrom, multipli mijelom), infekcijama (subakutni bakterijski endokarditis, tuberkuloza, sifilis), bolestima vezivnoga tkiva (reumatoidni artritis, sistemski eritematozni lupus) te nekim drugim bolestima i poremećajima (alkoholna bolest jetre, primjena kortikosteroida, stresni događaji).
Bazofilija se definira brojem bazofila u krvi u iznosu većem od 0,2 x 109/L, a može ju se naći kod bolesnika s bolestima vezivnoga tkiva, alergijskim i pseudoalergijskim reakcijama, idiopatskim crijevnim upalnim bolestima, malignim oboljenjima i drugim.
KRONIČNE MIJELOPROLIFERATIVNE BOLESTI
Uvod. Kronične mijeloproliferativne bolesti predstavljaju skupinu bolesti karakteriziranu poremećajem matične pluripotentne stanice koji rezultira pojačanim stvaranjem i nakupljanjem zrelih krvnih stanica u koštanoj srži i perifernoj krvi. U tu skupinu bolesti ubrajamo esencijalnu trombocitozu, policitemiju rubru veru, kroničnu primarnu mijelofibrozu te kroničnu mijeloičnu leukemiju. Osnovna im je karakteristika klonalna narav te mogućnost prelaska bolesti u sekundarnu mijelofibrozu. U nastavku će teksta biti opisane esencijalna trombocitoza, policitemija rubra vera, primarna mijelofibroza te kronična mijeloična leukemija.
ESENCIJALNA TROMBOCITOZA
Definicija. Esencijalna trombocitoza (idiopatska trombocitemija, primarna hemoragijska trombocitemija) predstavlja jednu od najčešćih kroničnih mijeloproliferativnih bolesti. Karakterizira ju prekomjerno stvaranje i nakupljanje trombocita. Iako se taj poremećaj većinom susreće u kasnijoj životnoj dobi (od petoga do sedmoga desetljeća života), može se pojaviti u bilo kojoj dobi, a današnja saznanja pokazuju nešto višu učestalost poremećaja kod žena u odnosu na muškarce (osobito ako se radi o esencijalnoj trombocitozi udruženoj sa specifičnim mutacijama). U oko 5 – 10 % slučajeva bolest može napredovati u neki drugi oblik kronične mijeloproliferativne bolesti, mijelodisplaziju ili akutnu leukemiju.
Etiopatogeneza. Esencijalna trombocitemija nastaje kao posljedica poremećaja multipotentne krvotvorne matične stanice koji rezultira pojačanom proliferacijom megakariocita u koštanoj srži i posljedičnim pojačanim stvaranjem trombocita. In vitro istraživanja pokazala su da je abnormalni klon megakariocita takvih bolesnika neovisan o djelovanju faktora rasta i da stanice pokazuju sposobnost autonomnoga dijeljenja, rasta i sazrijevanja. Točan uzrok i mehanizam nastanka poremećaja nije jasan, ali postoji izražena povezanost između određenih mutacija i pojave ove bolesti: JAK2-V617F mutacija (55 % slučajeva), CALR mutacija (36 % slučajeva) te MPL mutacija (4 % slučajeva). Karakteristično je da bolesnici koji imaju esencijalnu trombocitozu uz odsutnost tih mutacija imaju povoljniju prognozu.
JAK2 mutacija. JAK2 gen nalazi se na 9. kromosomu i odgovoran je za stvaranje i aktivnost tirozin kinaze vezane uz stanične receptore za citokine i faktore rasta. Posljedično mutaciji toga gena dolazi do prekomjerne konstitucijske aktivnosti navedenoga enzima, što za posljedicu ima staničnu hiperproliferaciju. Kod bolesnika s esencijalnom trombocitozom karakteristična je JAK2-V617F mutacija koja se može naći i kod drugih kroničnih mijeloproliferativnih bolesti (policitemija rubra vera, idiopatska mijelofibroza).
CALR mutacija. CALR gen se nalazi na 19. kromosomu, a odgovoran je za stvaranje CALR proteina koji je prisutan u endoplazmatskom retikulumu, jezgri i staničnoj membrani, a osim sudjelovanja u mehanizmima kontrole koncentracije kalcija, sudjeluje i u procesima stanične proliferacije i apoptoze. Do sada je opisano više od trideset vrsta mutacija toga gena povezanih s pojačanom staničnom proliferacijom te izbjegavanjem ulaska stanice u apoptozu.
MPL mutacija. MPL gen se nalazi na 1. kromosomu, a njegov genski produkt predstavlja transmembranski receptor odgovoran za vezanje trombopoetina koji regulira diferencijaciju megakariocita te nastanak trombocita. Mutacije toga gena povezane su s pojačanom osjetljivošću megakariocita na trombopoetin što rezultira njihovom pojačanom proliferacijom i produkcijom trombocita.
Klinička slika. Većina bolesnika s esencijalnom trombocitozom je asimptomatska te se bolest otkrije prilikom slučajno otkrivenoga patološkoga laboratorijskog nalaza povećanoga broja trombocita. Kliničke manifestacije esencijalne trombocitoze povezane su s povećanim rizikom od nastanka tromboza i krvarenja. Arterijske tromboze, posebice cerebralnih i koronarnih arterija, kod ovih su bolesnika češće od venskih tromboza te nastanak akutnoga infarkta miokarda ili cerebrovaskularnoga inzulta ponekad može biti prvi znak bolesti. Krvarenja se obično javljaju kod bolesnika s izrazito povećanim brojem trombocita (> 1000 – 1500 x 109/L), pa se i nazivaju paradoksalnim krvarenjima. Mogu se javiti spontano ili mogu biti izazvana traumama, a klasično se manifestiraju na koži, sluznicama i probavnom sustavu, ali mogu se javiti na bilo kojemu mjestu. U fizikalnom se statusu najčešće nađe umjerena splenomegalija, a kod težih oblika bolesti i eritromelalgija i livedo reticularis. Žene s esencijalnom trombocitozom sklonije su nastanku spontanih pobačaja. S obzirom na rizik od tromboze i krvarenja, bolesnike s esencijalnom trombocitozom dijelimo u dvije skupine: bolesnike s malim i s velikim rizikom, kako je prikazano u Tablici 6.15.
|
Tablica 6.15. Klinički rizici u bolesnika s esencijalnom trombocitozom
|
|
Bolesnici s malim rizikom
|
Bolesnici s velikim rizikom
|
|
Bolesnici mlađi od 60 godina;
Broj trombocita manji od 1000 x 109/L;
Negativna anamneza na krvarenje ili trombozu.
|
Bolesnici stariji od 60 godina;
i/ili
Pozitivna anamneza na krvarenje ili trombozu.
|
Dijagnoza esencijalne trombocitoze temelji se na laboratorijskim nalazima periferne krvi, nalazu biopsije koštane srži te isključivanju sekundarnih uzroka trombocitoze. Osnovni laboratorijski nalaz uključuje trombocitozu koja može biti različito izražena (> 450 x 109/L) s poremećenom morfologijom trombocita u razmazu periferne krvi (veliki trombociti koji međusobno stvaraju nakupine). Iako su vrijednosti eritrocita i leukocita većinom uredne, kod u dijela bolesnika mogu se naći umjerena anemija i leukocitoza. U uzorku koštane srži dominira hipercelularnost koja je obilježena proliferacijom megakariocitne loze (povećani broj velikih zrelih megakariocita) bez poremećaja granulocitopoeze i eritrocitopoeze. Za postavljanje konačne dijagnoze esencijalne trombocitoze trebaju biti zadovoljeni sva 4 velika kriterija ili 3 velika i 1 mali kriterij zadani prema Svjetskoj zdravstvenoj organizaciji (Tablica 6.16.).
|
Tablica 6.16. Dijagnostički kriteriji za esencijalnu trombocitozu (Svjetska zdravstvena organizacija)
|
|
Veliki kriteriji
|
- Prisutnost trajno povišenih vrijednosti trombocita i to u vrijednosti većoj od 450x109/L;
- Prisutnost proliferacije megakariocitne loze u uzroku biopsije koštane srži;
- Nepostojanje kriterija za policitemiju rubru veru, primarnu mijelofibrozu, BCR-ABL pozitivnu kroničnu mijeloičnu leukemiju, mijelodisplastični sindrom ili drugi mijeloidni zloćudni tumor;
- Dokaz JAK2V617F, CALR ili MLP mutacije.
|
|
Mali kriteriji
|
|
1. Prisutnost klonskih markera (abnormalan kariotip)
2. Odsutnost dokaza za reaktivnu trombocitozu
|
Diferencijalna dijagnoza. Osnovna diferencijalno-dijagnostička dvojba pri postavljanju dijagnoze esencijalne trombocitoze njezino je razlikovanje od sekundarne (reaktivne) trombocitoze koja se može pojaviti uslijed mnogih bolesti i stanja, kao što su akutna i kronična upala (reumatske bolesti, idiopatska crijevna upalna bolest), kronične infekcije, zloćudni tumori (metastatski karcinom, limfomi), benigna hematološka oboljenja (akutno krvarenje, akutna hemolitička anemija) kronična bolest bubrega (zatajenje bubrega, nefrotski sindrom), primjena lijekova (vinkristin, epinefrin, glukokortikoidi, ATRA, niskomolekularni heparin) i sl. Na sekundarnu trombocitozu ukazuje prisutnost mogućega uzročnog stanja ili bolesti, uredan nalaz biopsije koštane srži te uredna morfologija trombocita u razmazu periferne krvi.
Liječenje. Terapijski pristup bolesnicima s esencijalnom trombocitozom ponajprije ovisi o prisutnosti kliničkih simptoma te o riziku od nastanka tromboza i krvarenja. Svim se bolesnicima preporuča primjena antiagregacijske terapije, najčešće u obliku acetilsalicilne kiseline u dozi od 75 do 100 mg na dan. Specifično liječenje (citoredukcija) indicirana je kod svih simptomatskih bolesnika te kod asimptomatskih bolesnika s brojem trombocita većim od 1500 x 109/L ili prisutnim velikim rizikom od nastanka tromboza ili krvarenja. Citoredukcija se postiže primjenom hidroksiureje, anagrelida ili interferona alfa. U hitnim slučajevima liječenje se može provoditi i postupkom trombocitafereze.
Hidroksiureja predstavlja najdjelotvorniji lijek u liječenju ovih bolesnika. Točan mehanizam njezinoga djelovanja lijeka nije poznat, ali smatra se da dovodi do neposredne inhibicije sinteze molekule DNA. Učinak se vidi već za nekoliko dana, zbog čega je potrebno redovno pratiti krvnu sliku. Lijek se početno primjenjuje peroralno u dozi od 10 do 30 mg/kg/dan, a kasnije se za potrebe održavanja primjenjuju niže doze. Glavne su nuspojave anemija, leukopenija te sindrom lize tumora.
Anagrelid se koristi u drugoj liniji liječenja, kod bolesnika koji ne reagiraju na hidroksiureju ili razviju znatnu anemiju ili leukopeniju. On je selektivni inhibitor proliferacije i diferencijacije megakariocita, ali mu je terapijska primjena ograničena zbog čestih nuspojava u vidu srčanoga popuštanja, arterijske hipotenzije i srčanih aritmija uslijed vazodilatacijskoga i pozitivnoga inotropnog djelovanja. Liječenje počinje dozom od 2 mg/dan (podijeljeno u nekoliko dnevnih doza) uz postupno povećanje doze za 0,5 mg svakih sedam dana do postizanja učinka ili do maksimalne dnevne doze od 10 mg/dan.
Interferon alfa predstavlja jedan od najdjelotvornijih i najsigurnijih lijekova za postizanje smanjenja broja trombocita. ali mu učinak postaje vidljiv kroz mjesec dana. Primjenjuje se subkutano u dnevnoj dozi od 3 milijuna jedinica na dan. Najčešće su nuspojave simptomi nalik gripi, a prikladan je i za liječenje trudnica, s obzirom na teratogeni učinak hidroksiureje i nedovoljno ispitan učinak anagrelida na fetus.
Prognoza. Esencijalna trombocitoza najčešće je praćena kroničnim i benignim tijekom. Bez primjene liječenja godišnja stopa smrtnosti iznosi 1 %. Kod manje od 10 % bolesnika bilježi se transformacija bolesti u neki drugi oblik kronične mijeloproliferativne bolesti, mijelodisplaziju ili akutnu leukemiju.
POLICITEMIJA RUBRA VERA
Definicija. Policitemija rubra vera predstavlja kroničnu mijeloproliferativnu bolest karakteriziranu prekomjernim stvaranjem i nakupljanjem eritrocita u koštanoj srži i perifernoj krvi. Iako se može javiti u svakoj životnoj dobi, pojavnost policitemije rubre vere češća je nakon petoga desetljeća života, a procijenjena ukupna incidencija ove bolesti kreće se oko 2,5 na 100 000 stanovnika. Bolest pokazuje nešto češću pojavnost kod muškaraca u odnosu na žene.
Etiopatogeneza. Točan uzrok nastanka toga poremećaja do danas nije u potpunosti poznat. Osnovu bolesti čini poremećaj multipotentne krvotovorne matične stanice koji dominantno rezultira nekontroliranom proliferacijom stanica eritrocitopoeze, a u nešto manjoj mjeri i stanica granulocitopoeze i trombocitopoeze. Posljedično je u perifernoj krvi dominantno povećan broj eritrocita (eritrocitoza), dok su leukocitoza i trombocitoza umjerenoga tipa. Više od 95 % bolesnika ima ranije opisanu JAK2 mutaciju. Za razliku od esencijalne trombocitoze i primarne mijelofibroze gdje je prisutna samo JAK2V617F mutacija, bolesnici (iako u znatno manjem broju) imaju i drugi oblik JAK2 mutacije, a to je JAK2 ekson-12 mutacija. Stanice eritropoeze u koštanoj srži takvih bolesnika pokazuju sposobnost autonomne proliferacije i diferencijacije neovisno o prisutnosti eritropoetina i drugih faktora rasta. Eritropetin je kod bolesnika s policitemijom rubrom verom snižen. Također, stanice eritocitopoeze pokazuju i povećanu otpornost na apoptozu što je posredovano povećanom ekspresijom antiapoptotičnog proteina Bcl-x1.
Klinička slika. Klinička se obilježja policitemije rubre vere mogu podijeliti u dvije skupine: simptomi i znakovi koji su posljedica hiperviskoznosti i tromboembolijskih incidenata, te simptomi i znakovi koji su posljedica hipermetabolizma uslijed povećanoga broja stanica. Kao i kod esencijalne trombocitoze, policitemija rubra vera kod trudnica može biti povezana s povećanom sklonošću razvoju spontanih pobačaja.
Simptomi i znakovi hiperviskoznosti i tromboembolija su pletora (zagasito crvena boja kože i vidljivih sluznica kao posljedica kombinacije prepunjenih kapilara i povećane količine deoksigeniranoga hemoglobina), splenomegalija te arterijska hipertenzija, a bolesnici pokazuju povećanu sklonost razvoju venskih i arterijskih tromboza (češće su arterijske). Posljedično arterijskim trombozama nastaju cereborvaskularni inzult, akutni koronarni sindrom ili akutna ishemija ekstremiteta, a posljedično venskim trombozama mogu se razviti duboka venska tromboza gornjih ili donjih ekstremiteta, tromboza splanhičnih vena (vena porte, hepatalne vene) ili tromboza venskih kavernoznih sinusa. .
Simptomi i znakovi hipermetabolizma uključuju obilno znojenje, subfebrilne temperature, svrbež, hiperurikemiju (giht) te eritromelalgiju (sindrom koji se očituje crvenilom, toplinom i bolovima, obično donjih udova, izrazitije pri promjenama temperature).
Dijagnoza policitemije rubre vere postavlja se na osnovi kliničke slike, laboratorijskih nalaza periferne krvi te nalaza biopsije koštane srži. Temeljni laboratorijski nalazi uključuju povećanu vrijednost hemoglobina (više od 165 g/L za muškarce i više od 160 g/L za žene) i hematokrita (više od 49 % za muškarce i 48 % za žene) uz umjerenu trombocitozu i leukocitozu, a ostali laboratorijski nalazi koji upućuju na tu bolest su mikrocitoza (sniženi MCV), sideropenija, snižene vrijednosti eritropoetina, povišene vrijednosti vitamina B12 te hiperurikemija. Biopsija koštane srži ukazuje na izraženu hipercelularnost s dominacijom stanica eritroidne loze i manje izraženom proliferacijom granulocitne i trombocitne loze. U više od 95 % slučajeva nalaz za JAK2V617F mutaciju ili mutaciju JAK2 za ekson-12 je pozitivan. Pri postavljanju dijagnoze policitemije rubre vere koriste se kriteriji Svjetske zdravstvene organizacije (Tablica 6.17.).
|
Tablica 6.17. Dijagnostički kriteriji za policitemiju rubru veru
|
|
Veliki kriteriji
|
Mali kriteriji
|
- Hemoglobin > 165 g/L za muškarce ili 160 g/L za žene;
- Prisutnost JAK2 mutacije;
- Biopsija koštane srži s izraženom proliferacijom eritroidne stanične linije
- Snižene vrijednosti eritropetina u serumu.
|
|
|
Dijagnoza policitemije rubre vere izvjesna je ako su prisutna sva tri velika kriterija ili prva dva velika i mali kriterij.
|
Diferencijalna dijagnoza. Pristup bolesniku s eritrocitozom podrazumijeva najprije njezino razgraničenje na apsolutnu i relativnu eritrocitozu. Apsolutna eritrocitoza nastaje uslijed povećane mase eritrocita u perifernoj krvi što se može javiti kao primarni poremećaj (primarna eritrocitoza), kakav je policitemija rubra vera, ili kao sekundarni poremećaj (sekundarna eritrocitoza) koji se javlja kao odgovor na povećano stvaranje čimbenika koji stimuliraju eritroidnu lozu. Relativna eritrocitoza karakterizirana je povišenim vrijednostima hemoglobina i hematokrita koje nastaju kao posljedica smanjenja volumena plazme, a ne stvarnoga povećanja mase eritrocita. Temeljno razlikovanje tih kliničkih kategorija bazira se na određivanju vrijednosti eritropoetina koji je snižen pri policitemiji rubri veri, povišen u sekundarnoj eritrocitozi te uredan kod bolesnika s relativnom eritrocitozom. Ostale razlike prikazane su u Tablici 6.18.
|
Tablica 6.18. Diferencijalna dijagnoza eritrocitoze
|
|
Karakteristika
|
Policitemija rubra vera
|
Sekundarna erirocitoza
|
Relativna eritrocitoza
|
|
Ukupna masa eritrocita
|
Povećana
|
Povećana
|
Normalna
|
|
Leukocitoza
|
Prisutna
|
Nije prisutna
|
Nije prisutna
|
|
Trombocitoza
|
Prisutna
|
Nije prisutna
|
Nije prisutna
|
|
Eritropoetin
|
Snižen
|
Povišen
|
Normalan
|
|
Koštana srž
|
Panhiperplazija
|
Hiperplazija eritoidne loze
|
Normalna
|
|
Splenomegalija
|
Prisutna
|
Nije prisutna
|
Nije prisutna
|
Sekundarna eritrocitoza može nastati kao posljedica različitih bolesti i stanja, među kojima su: 1) maligni tumori koji luče eritropoetin ili njemu slične tvari (karcinom bubrega i jetre, hemangioblastom), 2) nemaligne bolesti praćene povećanom produkcijom eritropoetina (ciste bubrega, hidronefroza) te 3) hipoksično-anoksijska stanja koja izazivaju kompenzatorno lučenje eritropoetina (kronične plućne i srčane bolesti, kronične bolesti eritrocita). Relativna eritrocitoza nastaje kao posljedica deplecije intravaskularnoga volumena (dehidracija).
Liječenje. Osnovu liječenja čini smanjenje broja eritrocita, odnosno postizanje ciljnih vrijednosti hematokrita koje za muškarce iznose manje od 0,45 (45 %), a manje od 0,42 (42 %) za žene. Osnovu liječenja čini redovna primjena acetilsalicilne kiseline (sprječavanje trombotskih incidenata i paradoksalnoga krvarenja) te povremene venepunkcije. Kod težih oblika bolesti, tj. kod bolesnika kod kojih se kontrola bolesti ne postiže navedenim postupkom, kao i kod bolesnika s visokim rizikom od nastanka tromboza i krvarenja (bolesnici stariji od 60 godina s prisutnim kardiovaskularnim rizicima), indicirana je primjena mijelosupresivne terapije, odnosno citoredukcija.
Venepunkcija podrazumijeva evakuaciju venske krvi s ciljem redukcije mase eritrocita i ostalih stanica. Jednim se postupkom evakuira 400 do 500 mL krvi, a postupak se može primjenjivati svaka dva do četiri dana sve do postizanja ciljnih vrijednosti hematokrita, a nakon toga se postupak primjenjuje ovisno o laboratorijskim nalazima (obično svakih tri do šest mjeseci). Kod kroničnih srčanih bolesnika preporuča se evakuacija manje količine krvi po jednom postupku. Nakon postupka venepunkcije može se primijeniti određena količina kristaloidnih infuzijskih otopina kako bi se spriječio značajniji pad krvnoga tlaka (osobito ako se venepunkcija često izvodi). Glavna nuspojava učestalih venepunkcija uključuje razvoj sideropenije s posljedičnom mikrocitozom i hipokromijom.
Medikamentozna citoredukcija primjenjuje se kod bolesnika kod kojih se venepunkcijama ne postiže kontrola bolesti, definira se kao potreba za više od dvije mjesečne venepunkcije kako bi se održala zadovoljavajuća vrijednost hematokrita, kao i kod bolesnika s visokim rizikom od nastanka tromboza i krvarenja (bolesnici stariji od 60 godina s prisutnim kardiovaskularnim rizicima). Hidroksiureja je lijek prvoga izbora u liječenju ovih bolesnika, a primjenjuje se u dnevnoj dozi od 500 - 1500 mg/dan pazeći da se bolesnika ne uvede u trombocitopeniju ili leukopeniju. S obzirom na brzo i kratko djelovanje, hidroksiureja se kod većine bolesnika mora primjenjivati kontinuirano. Kod bolesnika koji ne odgovaraju na terapiju hidroksiurejom ili je ne toleriraju moguća je primjena ruksolitiniba, JAK2 inhibitora čiji su dobar učinak u liječenju ovih bolesnika pokazale dosada provedene studije, ili primjena pegeliranog interferona alfa. Primjena alkilirajućih kemoterapeutika, kao što su ranije primjenjivani busulfan ili radioaktivni fosfor, danas se ne preporuča zbog povezanosti s većim rizikom od progresije bolesti u akutnu leukemiju. Kod izražene trombocitoze koja ne reagira na hidroksiureju moguća je i primjena anagrelida.
Potporna terapija. Kod bolesnika s izraženim svrbežom mogu se primjenjivati antihistaminici (blokatori histaminskih H2 receptora) ili fototerapija, a kod bolesnika s hiperurikemijom indicirano je liječenje alopurinolom.
Prognoza. Policitemija rubra vera predstavlja bolest kroničnoga i indolentnog tijeka s medijanom preživljenja od oko petnaest godina. Glavni je uzrok mortaliteta bolesnika nastanak arterijskih tromboza (akutni infarkt miokarda, cerebrovaskularni inzult). S vremenom u 12 – 21 % bolesnika policitemija rubra vera prijeđe u mijelofibrozu ili kroničnu mijeloičnu leukemiju, a u oko 7 % slučajeva bilježi se i transformacija u akutnu mijeloičnu leukemiju praćenu lošom prognozom (ima slab terapijski odgovor).
PRIMARNA MIJELOFIBROZA
Definicija. Primarna (idiopatska) mijelofibroza označava kroničnu mijeloproliferativnu bolest karakteriziranu reaktivnom fibrozom koštane srži. Ona nastaje hiperplazijom abnormalnih megakariocita koji oslobađaju faktore koji stimuliraju fibroblaste. Kod nekih bolesnika policitemija rubra vera ili esencijalna trombocitoza u kasnijem tijeku bolesti može rezultirati razvojem fibroze koštane srži, što se naziva sekundarnom mijelofibrozom. Primarna mijelofibroza uglavnom je bolest starije populacije (uglavnom se javlja nakon petoga desetljeća života) s podjednakom učestalošću kod oba spola. Procijenjena učestalost iznosi 1 do 2 slučaja na milijun stanovnika.
Etiopatogeneza. Radi se o poremećaju matične hematopoetske stanice koji se u prvoj fazi bolesti (celularna faza) očituje proliferacijom svih staničnih loza (eritrocitne, granulocitne, trombocitne), pri čemu dominira hiperplazija abnormalnih, displastičnih megakariocita koji luče čimbenike koji stimuliraju fibroblaste (PDGF, engl. platelet-derived growth factor) posljedično čemu dolazi do pojačane produkcije kolagena koji formira kolagenske tračke i dovodi do postupne fibroze koštane srži. Umnoženi fibroblasti koji stvaraju kolagen nisu dio tumorskoga klona. Kako napreduje proces fibroze, tako postupno dolazi i do potiskivanja hematopoetskoga tkiva i smanjenja produkcije krvnih stanica (fibrozna faza bolesti), zbog čega se aktivira ekstramedularna hematopoeza i to dominantno u slezeni i jetri uslijed čega nastaju hepatosplenomegalija i pancitopenija. Kao i kod ostalih kroničnih mijeloproliferativnih bolesti, točan uzrok i mehanizam nastanka tih događaja nisu poznati. I kod tih se bolesnika mogu naći karakteristične JAK2 mutacije (JAK2V617F) te CALR i MPL mutacije. Također, oko 60 % bolesnika ima i specifične kromosomske aberacije, kao što su 9p, 20q-, 13q- te trisomija 8 ili 9.
Klinička slika. Bolesnici s primarnom mijelofibrozom dugo su asimptomatski te se dijagnoza često postavlja slučajno otkrivenim patološkim laboratorijskim nalazima. Početni simptomi su nespecifični i uključuju umor, malaksalost, mršavljenje i noćno preznojavanje, a oni su u prvom redu posljedica anemije i hipermetabolizma. Uslijed aktivacije ekstramedularne hematopoeze dolazi do razvoja izražene splenomegalije što se može manifestirati kao osjećaj tupoga bola i punoće u epigastriju i pod lijevim rebrenim lukom, a zbog nadražaja freničkoga živca mogu prisutni biti i bolovi u lijevom ramenu. Kod bolesnika su česti i infarkti slezene. Hepatomegalija je prisutna kod oko 50 % bolesnika s primarnom mijelofibrozom. Postupno, uslijed razvoja leukopenije i trombocitopenije, bolesnici postaju skloni infekcijama te razvijaju znakove hemoragijske dijateze i klinički manifestna krvarenja.
Dijagnoza se postavlja na osnovi laboratorijskih nalaza periferne krvi i nalaza biopsije koštane srži, a pozitivan nalaz JAK2V617F, CALR ili MPL mutacije podupiru njezino postojanje. U trenu postavljanja dijagnoze najčešće se primjećuje anemija s karakterističnim nalazom eritrocita u obliku suza (dakriociti) u razmazu periferne krvi, dok broj trombocita i leukocita može biti varijabilan (povišen, normalan ili snižen). Također, karakterističan je i bizaran izgled velikih i degranuliranih trombocita u razmazu periferne krvi. U kasnoj fazi bolesti prisutna je pancitopenija, kao i pojava nezrelih krvotvornih stanica u perifernoj krvi (leukoeritroblastoza). Dodatno se u laboratorijskim nalazima bilježi povišena razina laktat dehidrogenaze i hiperurikemija kao rezultat hipermetabolizma. Nalaz bioptata koštane srži ovisi o vremenu izvođenja biopsije: u celularnoj fazi dominira hipercelularnost koštane srži s proliferacijom svih usmjerenih mijeloidnih staničnih linija, dok u fazi fibroze nalazimo brojna retikulinska i koleganska vlakna koja potiskuju i zamjenjuju hematopoetsko tkivo, a nekada se može naći i osteoskleroza. Kriteriji Svjetske zdravstvene organizacije za postavljanje dijagnoze primarne mijelofibroze prikazani su u Tablici 6.19.
|
Tablica 6.19. Dijagnostički kriteriji za primarnu mijelofibrozu
|
|
Veliki kriteriji
|
Mali kriteriji
|
- Proliferacija i atipija megakariocita praćena retikulinskom ili kolagenskom fibrozom;
- Nepostojanje kriterija za policitemiju rubru veru, BCR-ABL pozitivnu kroničnu mijeloičnu leukemiju, mijelodisplastični sindrom i druge mijeloidne tumore;
- Dokaz JAK2V617F, CALR ili MPL mutacije ili prisutnost klonalnih biljega ili bez dokaza za reaktivnu fibrozu koštane srži.
- Leukoeritroblastoza;
- Povišena aktivnost laktat dehidrogenaze;
- Leukocitoza ≥ 11 × 109/L;
- Anemija;
- Splenomegalija.
|
|
Diferencijalna dijagnoza. Fibroza koštane srži može se naći u brojnim drugim bolestima, kao što su mijeloproliferativne i limfoproliferativne bolesti, metastatski karcinom te druge bolesti (bolesti vezivnoga tkiva, infekcije – tuberkuloza, osteomijelitis i sl.), na što treba misliti pri postavljanju dijagnoze i zbog toga se i treba voditi navedenim dijagnostičkim kriterijima Svjetske zdravstvene organizacije.
Liječenje. Specifičnoga liječenja primarne mijelofibroze nema. Inicijalno liječenje u početnoj fazi bolesti kada dominira mijeloproliferacija (leukocitoza, trombocitoza) čini primjena hidroksiureje (500 - 1000 mg/dan) uz korekciju anemije androgenima s ili bez kortikosteroida (prednison) te kontrolu splenomegalije (ruksolitinib, rijetko splenektomija i zračenje slezene). Kod bolesnika koji ne reagiraju na tu shemu liječenja u obzir dolaze alternativni lijekovi, kao što su intreferon alfa (indiciran je i kao osnovno liječenje trudnica) ili talidomid ili lenalidomid (lenalidomid je posebno učinkovit u liječenju primarne mijelofibroze u bolesnika s kromosomskom aberacijom 5q-). Kod bolesnika u uznapredovaloj fazi bolesti te bolesnika koji se ubrajaju u prognostički lošije skupine (kasnije opisano) liječenje se temelji na primjeni ruksolitiniba (JAK2 inhibitor) i/ili alogenoj transplantaciji koštane srži za koje se dokazano zna da poboljšavaju preživljenje.
Prognoza. Srednje preživljenje u bolesnika s postavljenom dijagnozom primarne mijelofibroze iznosi oko pet godina (u rasponu od 1 do 15 godina). Završna faza bolesti obilježena je generaliziranom astenijom, zatajenjem jetrene funkcije (hepatomegalija) i izraženoj sklonosti krvarenju, a moguća je i transformacija bolesti u akutnu mijeloičnu leukemiju. Nepovoljni prognostički čimbenici su starija životna dob (> 70 godina), niže vrijednosti hemoglobina (< 100 g/L), pojava nezrelih staničnih oblika u perifernoj krvi (> 2 % mijeloblasta, > 2 % eritronormoblasta), izražena leukocitoza (> 20 x 109/L), trombocitopenija (< 100 x 109/L), masivna splenomegalija te izraziti konstitucijski simptomi.
KRONIČNA MIJELOIČNA LEUKEMIJA
Definicija i epidemiologija. Kronična mijeloična leukemija (KML) klonalna je mijeloproliferativna bolest karakterizirana prekomjernom produkcijom zrelih stanica mijeloidne loze koje se potom nakupljaju u perifernoj krvi, a u više od 95 % slučajeva povezana je sa specifičnom kromosomskom promjenom - prisustvom Philadelphia kromosoma (Ph1 kromosom). Bolest se javlja isključivo u adultnoj populaciji, najčešće u petom desetljeću života s nešto češćom pojavnosti u muškaraca u odnosu na žene. Iako je KML obilježen kroničnim, sporoprogresivnim tijekom, kod dijela bolesnika moguć je nastup blastične krize ili transformacija bolesti u akutnu lekuemiju ili mijelofibrozu, što je ujedno povezano s lošijim ishodima i prognozom.
Etiopatogenza. Osim ionizirajućega zračenja, drugi, ranije spominjani rizični čimbenici koje se povezuje s nastankom hematoloških malignih bolesti nemaju dokazanu ulogu u nastanku ovoga oblika leukemije. KML se obično pojavljuje pet do deset godina nakon izlaganja ionizirajućem zračenju, što u prvom redu ovisi o dozi zračenja. Glavna kromosomska anomalija koju nalazimo u gotovo svim stanicama malignoga klona prisutnost je Philadelphia kromosoma. Radi se o balansiranoj, recipročnoj translokaciji genetskoga materijala između dugoga kraka kromosoma 9. i 22., pri čemu se onkogen ABL-1 (ranije znan kao c-ABL) s 9. kromosoma premješta na 22. kromosom u područje regije koja sadrži BCR gen (engl. breakpoint cluster region). Posljedično tome nastaje fuzijski BCR-ABL1 onkogen čiji genski produkt, BCR-ABL1 onkoprotein, posjeduje tirozin kinaznu aktivnost, odnosno ima sposobnost fosforilacije tirozina različitih unutarstaničnih proteina što čini ključni korak u njihovoj aktivaciji. Uslijed pojačane ekspresije BCR-ABL1 onkoproteina dolazi i do pojačane autofosforilacije unutarstaničnih proteina koji čine signalne puteve u aktivaciji gena koji potiču staničnu proliferaciju i dovode do inhibicije apoptoze, zbog čega se stanice s tom kromosomskom abnormalnošću pojačano dijele i umnožavaju te imaju produljeni životni vijek. Početna faza bolesti obilježena je pojačanim stvaranjem zrelih staničnih oblika (neutrofili, bazofili, eozinofili, monociti, megakariociti, eritroblasti i limfociti B) uz održanu funkciju koštane srži, stoga je i glavni nalaz kod ovih bolesnika periferna leukocitoza. Kod jednoga dijela bolesnika moguć je nastanak de novo mutacija ili kromosomskih poremećaja koji mogu dovesti do poremećaja sazrijevanja stanica mijeloične loze i njihovoga zadržavanja na razini nezrelih oblika, što se očituje razvojem blastične kroze ili transformacijom u akutnu leukemiju (u 70 % slučajeva radi se o akutnoj mijeloičnoj leukemiji, a u 30 % o akutnoj limfoblastičnoj leukemiji). Iako se kod najvećega broja bolesnika (> 90 %) citogenetskim i molekularnim tehnikama može dokazati prisutnost Philadelphia kromosoma i njegova genskog produkta - BCR-ABL1 onkoproteina, kod manjega broja bolesnika bolest može postojati i bez prisutnosti Philadelphia kromosoma (Ph1 negativna kronična mijeloična leukemija), pri čemu se kod dijela bolesnika može dokazati prisutnost BCR-ABL1 onkoproteina, dok se kod ostalih ne možemo dokazati niti njegova prisutnost, a prognostički gledano takvi bolesnici imaju teže oblike bolesti i lošije ishode.
Klinička slika. Kronična mijeloična leukemija ima dug asimptomatski period i većina se bolesnika dijagnosticira na temelju asimptomatskih patoloških laboratorijskih nalaza. Dominantno su izraženi simptomi pojačanoga staničnog metabolizma (umor, malaksalost, umjereno povišena temperatura, noćno preznojavanje, gubitak teka i pad na tjelesnoj težini) uz različito izraženu splenomegaliju koja je prisutna kod više od 90 % bolesnika. Rijetko se mogu javiti i simptomi hiperviskoznoga sindroma kod istražene leukocitoze (tromboembolijski incidenti, glavobolja, dispneja, prijapizam).
Prirodni tijek bolesti. U prirodnom tijeku bolesti razlikujemo tri stadija: kronična bolest, akcelerirana i blastična faza. Većina bolesnika s kroničnom mijeloičnom leukemijom nalazi se u kroničnoj fazi bolesti (> 90 %). Oni su većinom asimptomatski, a ukoliko ispoljavaju neke od simptoma, oni se brzo povlače nakon uvođenja terapije. Smrt je u toj fazi bolesti iznimno rijetka. Kod dijela bolesnika, osobito onih koji se ne liječe, bolest može ući u akceleriranu (ubrzanu) fazu. Tu fazu bolesti karakterizira pojava nezrelih staničnih oblika (10 – 20 % blasta u uzroku koštane srži i/ili > 15 % blasta u perifernoj krvi periferne krvi), a prate je i trombocitopenija, pogoršanje anemije te povećanje jetre i slezene (hepatosplenomegalija). Osim toga, u toj fazi bolesti se osim prisutnosti Philadelphia kromosoma može otkriti i prisutnost druge kromosomske aberacije, što se naziva klonalnom evolucijom. Uz današnju terapiju (inhibitori tirozin kinaze) godišnja učestalost prelaska kronične u akceleriranu fazu bolesti iznosi oko 2 %. Blastičnu fazu bolesti karakterizira prisutnost više od 20 % blasta u koštanoj srži i/ili perifernoj krvi ili razvijena ekstramedularna blastična bolest (pojava blasta u drugim organima i tkivima - slezena, limfni čvorovi, moždane ovojnice i sl.), a obično se razvija iz akcelerirane faze, iako se kod dijela bolesnika može razviti i bez nje. Kod gotovo svih bolesnika prisutne su neke od kromosomskih promjena uz postojeći Philadelphia kromosom (trisomija 8, izokromosom 17, dupliranje Ph1 i sl.). Bolesnici s blastičnom krizom imaju znatno lošije kliničke ishode.
Dijagnoza se postavlja na temelju laboratorijskih nalaza, citološkoga nalaza koštane srži te dokazom opisanoga citogenetskog i molekularnoga poremećaja.
Laboratorijski nalazi. Osnovni laboratorijski nalazi ukazuju na leukocitozu koja može biti različito izražena (od 10 do 300 x 109/L) uz dominaciju neutrofila u diferencijalnoj krvnoj slici, a povišeni su i bazofili i eozinofili. Broj blasta i promijelocita ne prelazi 20 % (obično ispod 5 %). Također, većina bolesnika ima i izraženu umjerenu normocitnu i normokromnu anemiju (hemoglobin niži od 110 g/L) te trombocitozu, dok je trombocitopenija rijetka i ukazuje na teži tijek bolesti i lošije ishode.
Citološki nalaz koštane srži. Koštana srž bolesnika je hipercelularna, pri čemu dominiraju stanice mijeloične loze te megakariociti. Također, poremećen je i odnos između granulocitnih stanica (stanice mijeloidne loze) i eritrocita, tzv. granulocitno-eritrocitni indeks koji kod takvih bolesnika iznosi 10 do 50:1, dok je normalni odnos 2 do 5:1. Udio blasta u staničnoj populaciji koštane srži je normalan ili tek blago povećan (o blastičnoj krizi govori se kada je udio blasta u koštanoj srži viši od 20 %). Fibrozu koštane srži razvije oko trećina bolesnika, ali se to obično vidi u kasnijim fazama bolesti i predstavlja negativan prognostički znak
Citogenetska i molekularna dijagnostika. Osnovni citogenetski nalaz bolesnika s KML-om uključuje prisutnost Philadelphia kromosoma (G-pruganje, FISH metoda), iako on ne mora biti prisutan kod svih bolesnika (< 5 % bolesnika). Molekularnim metodama (RT-PCR) dokazuje se prisutnost BCR-ABL1 onkoproteina koji se može dokazati kod 1 % bolesnika koji nemaju Philadelphia kromosom. Mjerenjem koncentracije BCR-ABL1 moguće je pratiti terapijski odgovor bolesnika s KML-om. Kod 10 do 15 % bolesnika moguće je uočiti i dodatne kromosomske promjene, kao što su gubitak Y kromosoma, trisomija 8, dvostruki Ph1 i sl.
Diferencijalna dijagnoza. Glavne diferencijalno dijagnostičke dileme povezane su s razlikovanjem KML-a od reaktivne leukocitoze povezane s infekcijom (mijeloidna leukemoidna reakcija) te drugih mijeloproliferativnih bolesti. U tom razlikovanju najznačajniju ulogu imaju citološka i molekularna dijagnostika, tj. dokazivanje prisutnosti Philadelphia kromosoma i BCR-ABL1 onkoproteina.
Liječenje. Osnovu liječenja čini suzbijanje malignoga BCR/ABL1 klona pri čemu glavnu ulogu imaju lijekovi koji djeluju kao inhibitori tirozin-kinaze te transplantacija alogene koštane srži koju se danas koristi samo uz određene indikacije. Kod bolesnika koji u trenutku postavljanja dijagnoze imaju sindrom leukostaze (hiperleukocitarni sindrom) opravdana je i terapija hidroksiurejom ili primjena postupka leukafereze. Modaliteti i preporuke za liječenje kronične mijeloične leukemije sažeti su u Tablici 6.20.
Utvrđivanje odgovora na liječenje. Danas se u utvrđivanju odgovora na liječenje koriste hematološki kriteriji (normalizacija kompletne i diferencijalne krvne slike), citogenetski kriteriji (smanjena ili odsutna prisutnost Philadelphia kromosoma u stanicama koštane srži) te molekularni kriteriji (kvantifikacija BCR/ABL1 prijepisa iz uzorka periferne krvi metodom RT-PCR). U pravilu se nakon započinjanja liječenja najprije postiže hematološki odgovor (unutar tri mjeseca), a potom citogenetski (tri do šest mjeseci) i molekularni odgovor (šest do dvanaest mjeseci). Bolesnici kod kojih se postignu ovi odgovori unutar godine dana imaju izvrsnu prognozu i dobre ishode, dok bolesnici kod kojih se ne postižu navedeni ciljevi, kod kojih izostane početno ostvaren terapijski odgovor ili steknu dodatnu mutaciju ili kromosomsku aberaciju imaju lošije ishode i u pravilu zahtijevaju promjenu terapijskoga pristupa.
Prva linija liječenja. U prvoj liniji liječenja danas se primjenjuju inhibitori tirozin kinaze prve generacije (imatinib) koji pokazuju dobar učinak u postizanju remisije bolesti i smanjenja stope prelaska bolesti u uznapredovalu fazu (akceleracija, blastična kriza). Iako inhibitori tirozin kinaze druge generacije (nilotinib i dasatinib) pokazuju bolju učinkovitost od imatiniba, ne koriste se svuda u prvoj liniji liječenja, već su rezervirani za bolesnike kod kojih se ne postigne terapijski uspjeh primjenom imatiniba.
Druga/treća linija liječenja. Promjena strategije liječenja indicirana je kod bolesnika kod kojih se primjenom navedenih lijekova prve linije ne postiže terapijski uspjeh, kao i bolesnika koji razviju intoleranciju ili rezistenciju na navedene lijekove ili imaju relaps bolesti. Kod takvih bolesnika na raspolaganju nam stoje novije generacije inhibitora tirozin kinaze kao što su nilotinib, dasatinib, bosutinib i ponatinib te omaketaksin (analog homoharingtonina, neselektivni inhibitor sinteze proteine, koji između ostalog smanjuje i vrijednosti BCR-ABL1 onkoproteina) i alogena transplantacija koštane srži. Primjena bosutiniba prakticira se kod bolesnika rezistentnih na tirozin kinazne inhibitore prve i druge generacije, osim kod bolesnika s dokazanom mutacijom T315I kod kojih je opravdana primjena ponatiniba, povezana s visokom stopom tromboembolijskih komplikacija. Primjena omaketaksina opravdana je kod bolesnika kod kojih postoji dokumentirana rezistencija na minimalno dva inhibitora tirozin kinaze.
Alogena transplantacija koštane srži. Taj način liječenja ovih bolesnika danas je rezerviran za bolesnike kod kojih se ne može postići zadovoljavajući terapijski odgovor unatoč medikamentnom liječenju, odnosno kod bolesnika kod kojih se pojavljuje progresija bolesti unatoč njihovoj primjeni.
Akcelerirana i blastična faza bolesti. Liječenje takvih bolesnika je agresivno. Osnovu čini primjena inhibitora tirozin kinaze u većim dozama nego kod bolesnika u kroničnoj fazi bolesti (prednost se daje lijekovima novije generacije) s ili bez primjene mijelosupresivne kemoterapije. Cilj takvoga medikamentnoznog liječenja je suprimirati maligni klon u što većoj mjeri te čim prije pristupiti alogenoj transplantaciji koštane srži.
|
Tablica 6.20. Modaliteti i preporuke za liječenje kronične mijeloične lekemije
|
|
Klinički scenarij
|
Terapija inhibitorima tirozin kinaze
|
Alogena transplantacija koštane srži
|
|
Akcelerirana ili blastična faza bolesti
|
Privremena terapija s ciljem što veće supresije bolesti, visoke doze lijekova
|
Preporuča se što ranije (čim je to moguće)
|
|
Kronična faza bolesti, neuspjeh imatiniba, prisutna T315I mutacija
|
Ponatinib kao privremena terapija, cilj što veća supresija bolesti
|
Preporuča se što ranije (osobito kod neuspjeha ponatiniba)
|
|
Kronična faza bolesti, neuspjeh imatiniba, bez mutacija, dobar inicijalni odgovor
|
Dugotrajna primjena tirozin kinaznih inhibitora druge generacije
|
Treća linija liječenja ako ne odgovara na tirozin kinazne inhibitore
|
|
Kronična faza bolesti, neuspjeh imatiniba i drugih inhibitora druge generacije
|
Privremena terapija inhibitorima treće generacije (ili omaketaksin), supresija bolesti
|
Primjenjuje se kao druga linija liječenja nakon medikamentne terapije
|
|
Kronična faza bolesti, neuspjeh imatiniba, starije osobe (> 70 god)
|
Dugotrajna primjena inhibitora druge generacije
|
Ovisno o kvaliteti i očekivanom trajanju života
|
Prognoza. Primjena inhibitora tirozin kinaze dovela je do značajnoga poboljšanja prognoze bolesnika. Ukupno gledano, više od 80 % bolesnika nema znakove progresije bolesti i ima zadovoljavajuću kvalitetu života kroz devet godina. Bolesnici s dobrim terapijskim odgovorom (postignuta hematološka, citogenetska i molekularna supresija bolesti u prvoj godini liječenja) imaju gotovo 100 %-tno devetogodišnje preživljenje, a za dobar dio bolesnika može se reći da su izliječeni. Bolesnici koji uđu u akceleriranu i blastičnu fazu bolesti imaju znatno lošije ishode (medijan preživljenja bolesnika s akceleriranom fazom iznosi pet godina, a s blastičnom fazom bolesti svega pet mjeseci).
MIJELODISPLASTIČNI SINDROM
Definicija. Mijelodisplastični sindrom (MDS) naziv je za skupinu klonalnih bolesti krvotvorne matične stanice koje su obilježene poremećenom diferencijacijom (sazrijevanje i morfologija) i proliferacijom jedne ili više loza uz karakterističnu povećanu i ubrzanu apoptozu stanica malignoga klona, zbog čega oboljeli razvijaju citopeniju u perifernoj krvi uz povećanu celularnost koštane srži. Pojačana apoptoza glavni je događaj koji razlikuje mijelodisplastični sindrom od akutnih leukemija jer onemogućava nakupljanje stanica malignoga klona u perifernoj krvi. Ipak, značajan broj bolesnika s mijelodisplastičnim sindromom razvije akutnu leukemiju (obično akutnu mijeloičnu), pa se taj sindrom nekada naziva i sindrom preleukemije.
Epidemiologija. Mijelodisplastični sindrom danas se ponajprije smatra bolešću starije populacije, s obzirom na to da se njegova učestalost znatno povećava nakon petoga desetljeća života, no može se javiti i kod mlađih osoba, osobito u slučaju izloženosti nekima od snažnih rizičnih čimbenika, kao što su ionizirajuće zračenje ili primjena citostatika (alkilirajući lijekovi, inhibitori topoizomeraze II). Pojavnost mijelodisplastičnoga sindroma nakon izlaganja ionizirajućem zračenju ili nakon primjene alkilirajućih lijekova obično se zbiva nakon vremenskoga razdoblja od pet do deset godina, dok je taj period kraći nakon primjene inhibitora topoizomeraze i iznosi od jedne do tri godine. Procijenjena incidencija u našim krajevima iznosi 2 do 10 slučajeva na 100 000 stanovnika, ali se može povećati i do 50 na 100 000 kod osoba starijih od 70 godina. Bolest se nešto češće javlja kod muškaraca u odnosu na žene.
Etiologija. Kao i kod akutnih leukemija, točan uzrok i mehanizam razvoja ovoga poremećaja nije jasan. Kao jedan od najznačajnijih rizičnih čimbenika navodi se sam proces starenja jer dovodi do posljedične akumulacije somatskih mutacija koje mogu rezultirati malignom pretvorbom krvotvorne matične stanice. Ostali rizični čimbenici koje se povezuje s razvojem mijelodisplastičnoga sindroma su izlaganje ionizirajućem zračenju, primjena alkilirajućih kemoterapeutika (klorambucil, mefalan) i inhibitora topoizomeraze (etpozid, doksorubicin), kao i izloženost drugim kemikalijama (petrolej, pesticidi i herbicidi). Obiteljska pojavnost mijelodisplastičnoga sindroma je rijetka, a povezuje se s mutacijama RUNX1 i GATA2 gena.
Patogeneza. Osnovni poremećaj u mijelodisplastičnom sindromu označava malignu transformaciju jedne od matičnih stanica usmjerenih loza uslijed stečene (rijetko nasljedne) mutacije nekih od gena koji upravljaju staničnom diferencijacijom i proliferacijom. Navedene mutacije rezultiraju poremećenom funkcijom tumor-supresorskih gena ili pojačanom ekspresijom staničnih onkogena posljedično čemu dolazi do poremećaja u sazrijevanju i morfologiji stanica malignoga klona koje pri tome potiskuju normalno hematopoetsko tkivo koštane srži zbog čega se i razvija njezina disfunkcija. U početnoj fazi stanice malignoga klona ubrzano propadaju u samoj koštanoj srži uslijed pojačane apoptoze, a unatoč povećanoj celularnosti koštane srži u perifernoj je krvi prisutna citopeniju što predstavlja glavno obilježje bolesti. Kod dijela bolesnika s progresijom bolesti pod utjecajem nakupljanja dodatnih mutacija sa slabljenjem apoptoze jedan klon počinje dominirati posljedično čemu dolazi i do povećanja broja tih stanica, što u konačnici označava prijelaz bolesti prema akutnoj leukemiji.
Citogenetski poremećaji. Iako je pojava mijelodisplastičnoga sindroma snažno povezana sa stečenim genetskim promjenama, niti jedna od njih ne dominira, a poznato je više od 25 takvih mutacija. Najčešće su mutacije SF3B1 (20 – 25 %), TET2 (20 %), ASXL1 (10 – 20 %), DNMT3A (10 – 15 %), RUNX1 (10 – 15 %), U2AF1 (5 – 10 %), SRSF2 (5 - 10%), TP53 (5 – 10 %) i dr. Kod više od 90 % bolesnika s mijelodisplastičnim sindrom moguće je detektirati barem jednu somatsku mutaciju, a često je istodobno prisutno više njih. Poremećaji kromosoma prisutni su kod oko polovine bolesnika, a najčešće se radi o deleciji dijelova ili cijelih kromosoma 5, 7 ili 20, te o trisomiji kromosoma 8 i translokaciji 11q23 kromosoma i njegovoj deleciji. Citogenetske kategorije u bolesnika s mijelodisplastičnim sindromom prikazane su u Tablici 6.21.
|
Tablica 6.21. Citogenetske kategorije u bolesnika s mijelodisplastičnim sindromom
|
|
Citogenetska kategorija
|
Citogenetski nalaz
|
|
Vrlo dobra
|
-Y, del(11q)
|
|
Dobra
|
Normalni kariogram, del(5q), del(12p), del(20q)
|
|
Srednja
|
del(7q), +8, +19, i(17q)
|
|
Loša
|
-7, inv(3)/t(3q)/del(3q), -7/del(7q)
|
|
Vrlo loša
|
Složene s više od 3 abnormalnosti
|
Klasifikacija. Službenu klasifikaciju mijelodisplastičnoga sindroma dala je Svjetska zdravstvena organizacija 2008. godine. Ona ovaj sindrom dijeli u nekoliko kliničkih entiteta: 1) refraktorna citopenija s displazijom jedne loze (anemija, neutropenija ili trombocitopenija), 2) refraktorna anemija s prstenastim sideroblastima, 3) mijelodisplastični sindrom vezan uz izoliranu deleciju kromosoma 5 - del(5q), 4) refraktorna citopenija s višeloznom displazijom, 5) refraktorna anemija s viškom blasta 1, 6) refraktorna anemija s viškom blasta 2 te 7) neklasificirani mijelodisplastični sindrom. Zbog eksponencijalnoga rasta broja informacija i novih saznanja o genetici MDS-a, Svjetska zdravstvena organizacija revidirala je postojeću klasifikaciju i 2016. godine donijela novu, koja u obzir uzima nekoliko obilježja - kao što su jednolinijske ili multilinijske displazije, prstenaste sideroblaste, suvišak blasta te citogenetičku abnormalnost del( 5q) – Tablica 6.23. Kako je klasifikacija iz 2016. godine relativno nova, danas je još uvijek u upotrebi klasifikacija iz 2008.
Klinička slika. Bolest karakterizira relativno dug asimptomatski period, pa se većina bolesnika dijagnosticira obradom slučajno otkrivene anemije u laboratorijskim nalazima. Prve i osnovne kliničke manifestacije mijelodisplastičnogs sindroma su anemijski sindrom koji obuhvaća umor, malaksalost, brzo zamaranje, palpitacije te osjećaj nestašice zraka uz zapaženo bljedilo i smanjenu prokrvljenost kože i sluznica. Kod razvijene trombocitopenije javlja se hemoragijska dijateza (produljena i teško zaustavljiva krvarenja, kožna krvarenja, spontana krvarenja), a uslijed leukopenije bolesnici su skloni infekcijama, osobito bakterijskim. Kod progresije bolesti i transformacije u akutnu leukemiju bolesnici mogu imati i hepatosplenomegaliju.
Dijagnoza mijelodisplastičnoga sindroma postavlja se na temelju laboratorijskih nalaza periferne krvi te nalaza biopsije/punkcije koštane srži koja obuhvaća citološku, citogenetsku i molekularnu te histološku analizu.
Laboratorijski nalazi. Jedan od najkarakterističnijih laboratorijskih nalaza je nalaz makrocitne anemije kod starijih osoba koju se ne može povezati s deficitom vitamina B skupina i alkoholizmom, osobito ako je udružena s trombocitopenijom i/ili leukopenijom, odnosno neutropenijom. Anemiju je prisutna kod više od 85 % bolesnika, neutropenija kod oko 50 % bolesnika, a trombocitopeniju kod oko 25 % bolesnika. U razmazu periferne krvi eritrociti su obično ovalni, a neutrofili i trombociti hipogranulirani.
Analiza koštane srži. Koštana srž bolesnika obično je hipercelularna, rjeđe uredna ili hipocelularna. Ovisno o zahvaćenosti staničnih loza, možemo naći različite morfološke promjene stanica: eritroidna loza (megaloblasti, prstenasti sideroblasti, internuklearni mostovi, karioreksa, fragmentacija jezgre, vakuolizacija citoplazme), granulocitna loza (hipogranulacija stanica, zastoj sazrijevanja na razini mijelocita) te megakariocitna loza (mikomegakariociti, hipogranulacija, povećan broj jezgara). Citogenetskom se analizom mogu uočiti različite kromosomske promjene kod 40 do 70 % bolesnika, na primjer delecije dugih krakova kromosoma 5, 7, 11, 13 i 20, delecije kratkih krakova kromosoma 12 i 13, trisomija kromosoma 8, 11, 21 i dr. Molekularnim se metodama mogu otkriti mutacije različitih gena, kao što su SF3B1, TET2, SRSF2, ASXL1, DNMT3A, RUNX1, U2AF1, TP53, EZH2 i dr.
Diferencijalna dijagnoza. Mijelodisplastični sindrom potrebno je razlikovati od megaloblastične anemije, aplastične anemije, mijelofibroze, pancitopenije uzrokovane HIV infekcijom te utjecaja lijekova. Razlika između akutne mijeloične leukemije i mijelodisplastičnoga sindroma temelji se na udjelu nezrelih staničnih oblika (blasta) koji u akutnoj leukemiji prelazi 20 %.
Prognostički bodovni sustav. Najčešće korišten i općeprihvaćen prognostički bodovni sustav za bolesnike s mijelodisplastičnim sindrom je revidirani IPSS sustav (IPSS-R, engl. Revised International Prognostic Scoring System for Myelodysplasia) koji se temelji na bodovanju osnovnih karakteristika bolesti: citogenetska kategorija, postotak blasta u koštanoj srži te vrijednosti hemoglobina, trombocita i neutrofila (Tablica 6.22.).
|
Tablica 6.22. IPSS-R bodovni sustav
|
|
Bod
|
Razred kariotipa
|
Blasti u koštanoj srži (%)
|
Hemoglobin (g/dL)
|
Trombociti (x109/L)
|
Neutrofili (x109/L)
|
|
0
|
Jako dobar
|
≤ 2
|
≥ 10
|
≥ 100
|
≥ 0.8
|
|
0.5
|
|
|
|
50 - 99
|
< 0.8
|
|
1.0
|
Dobar
|
2.1 - 4.9
|
8 - 9.9
|
< 50
|
|
|
1.5
|
|
|
< 8
|
|
|
|
2.0
|
Intermedijaran
|
5 - 10
|
|
|
|
|
3.0
|
Loš
|
> 10
|
|
|
|
|
4.0
|
Vrlo loš
|
|
|
|
|
|
Rizične kategorije: vrlo mala (< 1.5 bodova), mala (2.0 - 3.0 boda), srednja (3.5 - 4.5 boda), visoka (5.0 - 6.0 bodova) te vrlo visoka (> 6 bodova).
|
Liječenje. Glavni terapijski ciljevi kod bolesnika s mijelodisplastičnim sindromom su kontrola simptoma i znakova citopenije te smanjenje progresije u akutnu mijeloičnu leukemiju. Liječenje je indicirano za sve simptomatske bolesnike, a temelji se na individualnom riziku, odnosno dobi i kondicijskom stanju bolesnika te IPSS-R prognostičkom sustavu prema kojemu bolesnike svrstavamo u dvije kategorije: bolesnici s niskim rizikom (< 3 boda) i bolesnici s visokim rizikom (> 3.5 boda).
Niskorizični bolesnici. Osnovu liječenja mijelodisplastičnoga sindroma kod ove skupine bolesnika čini korekcija citopenije. Anemija se kao glavna klinička manifestacija korigira primjenom eritropoetina (epoetin) koji značajno povisuje hematokrit te smanjuje potrebu za učestalim transfuzijama, osobito ako se uz njega povremeno primjenjuje i faktor stimulacije granulocitnih kolonija (G-CSF). Kod bolesnika koji zahtijevaju učestale transfuzije koncentiranih eritrocita treba primjenjivati i vezače željeza (npr. deferasiroks) kako bi se izbjeglo preopterećenje željezom. Kod bolesnika s izraženom neutropenijom primjenjuje se filgrastim, dok se kod bolesnika s trombocitopenijom primjenjuju oralni (eltrombopag) ili subkutani trombopoetinski analozi (romiplostim). Kod bolesnika s delecijom del(5q) dobri rezultati postižu se primjenom imunomodulatora lenalidomida.
Visokorizični bolesnici i bolesnici koji ne reagiraju na terapiju. Kod bolesnika koji ne reagiraju na opisano liječenje, kao i kod visokorizičnih bolesnika najbolji rezultati postižu se alogenom transplantacijom krvotvornih matičnih stanica, ako su bolesnici dobno i kondicijski pogodni. Kod ostalih su bolesnika na raspolaganju hipometilirajući kemoterapeutici (lijekovi koji inhibiraju metilaciju molekule DNA), kao što su azacitidin ili decitabin, a prednost se daje azacitadinu koji se može koristiti zajedno s lenalidomidom. Primjena kemoterapeutika i/ili alogena transplantacija dokazano smanjuju mogućnost prelaska mijelodisplastičnoga sindroma u akutnu mijeloičnu leukemiju.
Prognoza. Prirodni tijek mijelodisplastičnoga sindroma pokazuje znatne razlike između pojedinih bolesnika na način da jedni imaju kroničnu stabilnu bolest, dok se kod drugih pojavljuje brza progresija u akutnu mijeloičnu leukemiju. Preživljenje, kao i stopa transformacije u akutnu leukemiju ovisi o prognostičkoj skupini prema IPSS-R kako je to navedeno u Tablici 6.22. Načelno gledano, većina bolesnika umire od izražene neutropenije i trombocitopenije (infekcije ili krvarenje) ili od posljedica transformacije u akutnu leukemiju.
|
Tablica 6.23. Klasifikacija mijelodisplastičnog sindroma prema SZO (2016.)
|
|
Naziv
|
Displastične linije
|
Citopenije
|
Prstenasti sideroblasti/ % KS eritoidnih elemenata
|
Broj blasta u koštanoj srži i perifernoj krvi
|
Citogenetika i kariotip
|
|
MDS s jednolinijskom displazijom (MDS-SLD)
|
1
|
1 ili 2
|
< 15 %/ < 5%
|
KS < 5%, PK < 1%
Bez Aurerovih štapića
|
Bilo koji, osim ako ne zadovoljava kriterije za MDS s del(5q)
|
|
MDS s višelinijskom displazijom (MDS-MLD)
|
2 ili 3
|
1 - 3
|
< 15 %/ < 5 %
|
KS < 5 %, PK < 1 %
Bez Aurerovih štapića
|
Bilo koji, osim ako ne zadovoljava kriterije za MDS s del(5q)
|
|
MDS s prstenastim sideroblastima (MDS-RS)
|
|
|
|
|
|
|
MDS-RS s jednolinijskom displazijom (MDS-RS-SLD)
|
1
|
1 ili 2
|
≥ 15 %/ ≥ 5 %
|
KS < 5 %, PK < 1 %
Bez Aurerovih štapića
|
Bilo koji, osim ako ne zadovoljava kriterije za MDS s del(5q)
|
|
MDS-RS s višelinijskom displazijom (MDS-RS-MLD)
|
2 ili 3
|
1 - 3
|
≥ 15 %/ ≥ 5 %
|
KS < 5 %, PK < 1 %
Bez Aurerovih štapića
|
Bilo koji, osim ako ne zadovoljava kriterije za MDS s del(5q)
|
|
MDS s izoliranom del(5q)
|
1 - 3
|
1 - 2
|
Nema ili bilo koji
|
KS < 5 %, PK < 1 %
Bez Aurerovih štapića
|
del(5q) sama ili jedna dodatna abnormalnost, osim -7 ili del(7q)
|
|
MDS sa suviškom blasta (MDS-EB)
|
|
|
|
|
|
|
MDS-EB-1
|
0 - 3
|
1 - 3
|
Nema ili bilo koji
|
KS 5 – 9 %
PK 2 – 4 %
Bez Aurerovih štapića
|
Bilo koji
|
|
MDS-EB-1
|
0 - 3
|
1 - 3
|
Nema ili bilo koji
|
KS 10 – 19 %
PK 5 – 19 %
Ili Aurerovi štapići
|
Bilo koji
|
|
MDS neklasificirani (MDS-U)
|
|
|
|
|
|
|
S 1 % blasta u krvi
|
1 - 3
|
1 - 3
|
Nema ili bilo koji
|
KS < 5 %, PK = 1 %
Bez Aurerovih štapića
|
Bilo koji
|
|
S jednolinijskom displazijom i pancitopenijom
|
1
|
3
|
Nema ili bilo koji
|
KS < 5 %, PK < 1 %
Bez Aurerovih štapića
|
Bilo koji
|
|
Definirajuća citogenetička abnormalnost
|
0
|
1 - 3
|
< 15 %
|
KS < 5 %, PK < 1 %
Bez Aurerovih štapića
|
MDS definirajuća abnormalnost
|
|
Refraktorna citopenija u djetinjstvu
|
1 - 3
|
1 - 3
|
Nema
|
KS < 5 %, PK < 2 %
|
Bilo koji
|
MALIGNE HEMATOLOŠKE BOLESTI
Maligne hematološke bolesti nastaju zloćudnom transformacijom stanica mijeloidnoga ili limfoidnog sustava koja može nastati na bilo kojoj razvojnoj razini, od matične hematopoetske stanice preko nezrelih oblika mijeloidne ili limfoidne stanične linije pa sve do zrelih krvnih stanica. Podjela hematoloških malignih bolesti je kompleksna i uključuje više kategorija (mjesto nastanka bolesti, stanično porijeklo, kliničko ponašanje, citogenetska obilježja i sl.).
Hematološke maligne bolesti obuhvaćaju tri velike skupine bolesti, a to su leukemije, limfomi i plazmastanične neoplazme. Također, u maligne hematološke bolesti ubrajaju se i kronične mijeloproliferativne bolesti te sindrom mijelodisplazije koji su zbog svojih kliničkih obilježja zasebno opisani. Leukemije su maligne hematološke bolesti koje primarno nastaju u koštanoj srži, a potom invadiraju perifernu krv i druge organe, dok limfomi predstavljaju maligne hematološke bolesti koje primarno nastaju u limfnim tkivima (limfni čvorovi, limfna tkiva pridružena organima), a potom invadiraju perifernu krv i druge organe. Plazmastanične neoplazme posebno su izdvojene hematološke maligne bolesti koje nastaju iz plazma stanica, a predstavnik im je multipli mijelom (plazmacitom).
S obzirom na stanično porijeklo, maligne hematološke bolesti mogu porijeklom biti od stanica mijeloidne loze iz kojih nastaju granulociti, monociti, eritrociti i trombociti ili mogu nastati od stanica limfoidne loze iz kojih nastaju T i B-limfociti. Stanice mijeloidne loze nalaze se samo u koštanoj srži, pa se ove maligne hematološke bolesti prezentiraju kao leukemije, dok se stanice limfoidne loze mogu nalaziti i u koštanoj srži i u limfnim tkivima, pa se mogu prezentirati ili kao leukemija ili kao limfom.
Kliničke karakteristike malignih hematoloških bolesti ponajprije ovise o zrelosti stanica u kojima nastaje zloćudna transformacija. Ukoliko zloćudna transformacija nastaje na razini zrelih, diferenciranih stanica nastaju maligne neoplazme koje obilježava povoljniji klinički tijek i spora progresija (niski stupanj proliferacije), pa takve leukemije nazivamo kroničnima, a limfome indolentnima. Nasuprot tome, ako zloćudna transformacija nastane na razini nezrelih, prekursorskih (nediferenciranih) stanica, razviju se maligne neoplazme koje obilježava nepovoljniji klinički tijek i brža progresija (visoki stupanj proliferacije) te takve leukemije nazivamo akutnima, a limfome agresivnima ili vrlo agresivnima.
Iako su općenito gledano maligne hematološke bolesti bolesti starije populacije, postoje specifični oblici koji se javljaju isključivo u mlađoj životnoj dobi. Također je karakteristično da se akutni, odnosno agresivni oblici bolesti češće javljaju u mlađih osoba, dok se kronični/indolentni oblici češće javljaju u starijih bolesnika.
Etiopatogeneza malignih hematološkh bolesti nije u potpunosti jasna, ali se zna da njihova pojavnost može biti povezana s nekim virusima, izlaganjem kemijskim i drugim toksičnim tvarima (lijekovi) ili ionizirajućem zračenju. Osnovni pretpostavljeni mehanizam nastanka ovih bolesti podrazumijeva nastanak unutarstaničnoga poremećaja u procesu kontrole proliferacije ili apoptoze stanice, što rezultira njihovim prekomjernim umnažanjem te povećanjem ukupne stanične mase. Pri tome veliku ulogu imaju specifične kromosomske aberacije i genetske mutacije. One ne samo da imaju ulogu u rasvjetljivanju i shvaćanju mehanizma nastanka bolesti, nego imaju i praktičnu primjenu zbog povezanosti s terapijskim odgovorom i prognozom.
Leukemije predstavljaju maligne hematološke bolesti karakterizirane malignom transformacijom i proliferacijom stanica mijeloidne ili limfoidne loze koja primarno nastaje u koštanoj srži, a potom se širi u perifernu krv i druge organe (jetra, slezena, limfni čvorovi). Ovisno o vrsti stanica iz koje potječu i kliničkim obilježjima, razlikuju se četiri temeljna oblika leukemije: akutna mijeloična leukemija (AML), akutna limfoblastična leukemija (ALL), kronična mijeloična leukemija (KML) te kronična limfocitna leukemija (KLL). Već se iz samoga naziva bolesti može iščitati da akutne leukemije nastaju iz nezrelih staničnih oblika (blasti), dok kronične leukemije nastaju iz zrelih, diferenciranih staničnih oblika.
Procijenjena ukupna godišnja učestalost leukemija u općoj populaciji iznosi oko 10 slučajeva na 100 000 stanovnika. Leukemije se nešto češće javljaju kod muškaraca u odnosu na žene, i to u odnosu 3:2 kod akutnih leukemija i 2:1 kod kroničnih leukemija. Iako se akutne leukemije mogu javiti u bilo kojoj životnoj dobi, nešto im je viša učestalost kod djece i mlađih odraslih osoba, dok se pojavnost kroničnih leukemija uglavnom povezuje sa starijom životnom dobi.
Iako je uzrok nastanka i razvoja leukemija nepoznat, njihova se pojavnost obično povezuje s genetskim i okolišnim faktorima kao što su zračenje, kemikalije i lijekovi te virusi. Povezanost ionizirajućega zračenja s pojavom leukemija dobro je istražena na životinjskim eksperimentalnim modelima, a jasna je i pozitivna korelacija između pojavnosti leukemije te izloženosti ionizirajućem zračenju u terapijske svrhe. Od kemijskih agenasa koji se povezuju s razvojem leukemije najčešće se izdvaja benzen, a od lijekova alkilirajući kemoterapeutici (melfalan) te inhibitori topoizomeraze II (etopozid), i to ponajviše s pojavom akutne mijeloične leukemije. Od virusa se s razvojem leukemije najčešće povezuje infekcija humanim T-leukemijskim virusom tipa 1 (HTLV-1), što je posebno izraženo u Japanu te nekim dijelovima Amerike i Afrike.
Iako je zamijećena veća pojavnost leukemija između identičnih blizanaca, kod bolesnika s određenim genetskim sindromima (Downov sindrom, Klinefelterov sindrom, Wiskott-Aldrichov sindrom), kao i češća pojavnost unutar nekih obitelji, genetsku osnovu leukemija u prvom redu čine stečene somatske mutacije koje pogađaju gene čiji genski produkti sudjeluju u kontroli stanične diferencijacije, proliferacije i apoptoze. Te mutacije mogu biti povezane s jasno uočljivim kromosomskim promjenama, ali se mogu javiti i bez njih. Pojedine karakteristične kromosomske aberacije i genske mutacije bit će opisane u odgovarajućim dijelovima teksta.
Zloćudna transformacija stanice limfoidne ili mijeloidne loze rezultirat će prekomjernim umnažanjem i proliferacijom, pri čemu se povećava ukupna tumorska masa. U leukemiji se to primarno zbiva na razini koštane srži pri čemu tumorske stanice potiskuju i infiltriraju normalno hematopoetsko tkivo čime se potiskuje i smanjuje proizvodnja ostalih krvnih stanica, zbog čega se uz leukocitozu razvija anemija i trombocitopenija. Postupno tumorske stanice iz koštane srži dospijevaju i u perifernu krv, a putem periferne krvi mogu invadirati i ostala tkiva, poglavito jetru, slezenu i limfne čvorove. Zbog velike količine stanica razvija se hipermetaboličko stanje koje nazivamo hiperleukocitarni sindrom. Uz povećano stvaranje stanica, prisutna je i povećana njihova razgradnja, zbog čega dolazi do porasta vrijednosti laktat-dehidorgenaze, hiperurikemije, hiperkalijemije i metaboličke acidoze. Povećan stanični udio u krvi (u odnosu na plazmu) očituje se povećanjem vrijednosti hematokrita i hiperviskoznosti, zbog čega su bolesnici skloni razviju trombotskih događaja.
Uvod. Akutne leukemije predstavljaju skupinu hematoloških malignih bolesti koje nastaju malignom transformacijom nezrele (neusmjerene ili dijelom usmjerene) stanice limfoidne ili mijeloidne loze, prema čemu se akutne leukemije i dijele u dvije velike skupine: akutne mijeloične leukemije i akutne limfoblastične leukemije. U odrasloj su populaciji akutne mijeloične leukemije i do četiri puta češće u odnosu na akutne limfoblastične leukemije, a učestalost im raste s dobi. Temeljno zbivanje u akutnim leukemijama podrazumijeva proliferaciju malignoga klona unutar koštane srži, koji potiskuje i zamjenjuje normalno hematopetsko tkivo (razvoj pancitopenije), a potom i prisutnost malignih stanica u perifernoj krvi i drugim organima (jetra, slezena, limfni čvorovi, koža, probavni sustav i sl.).
AKUTNA MIJELOIČNA LEUKEMIJA
Definicija. Akutna mijeloična leukemija (AML) predstavlja akutnu leukemiju koja nastaje malignom transformacijom, proliferacijom i nakupljanjem nezrelih stanica mijeloidne loze najprije unutar koštane srži, a potom u perifernoj krvi te drugim organima.
Epidemiologija. Učestalost AML-a iznosi 2 do 2,5 novootkrivena slučaja na 100 000 stanovnika. Iako učestalost te leukemije raste sa životnom dobi, najčešća je njezina pojavnost u šestom desetljeću života.
Etiopatogeneza. Kao i u drugim oblicima leukemija, točan uzrok i mehanizam nastanaka bolesti nije poznat. Kao rizični čimbenici razvoja navode se genetska predispozicija, izlaganje zračenju i toksinima, infekcije onkogenim virusima te primjena različitih lijekova. Osnovni pretpostavljeni mehanizam nastanka akutne mijeloične leukemije (kao i drugih oblika leukemija) uključuje genetske promjene u smislu aktivacije protoonkogena i/ili inaktivacije tumor supresorskih gena što rezultira gubitkom kontrole nad staničnim umnažanjem i posljedičnom hiperproliferacijom, čime od jedne maligno promijenjene stanice nastaje cijeli klon malignih stanica, a to je uvjetovano kromosomskim translokacijama, točkastim mutacijama ili inaktivacijom ili amplifikacijom gena. Maligni klon postupno potiskuje normalno hematopoetsko tkivo zbog čega se postupno razvijaju anemija i trombocitopenija. S progresijom bolesti leukemične stanice iz koštane srži dospijevaju i u perifernu krv te se mogu nakupljati i u drugim tkivima i organima.
Patogenetska obilježja. Akutnu mijeloičnu leukemiju karakteriziraju određena morfološka (izgled leukemijskih stanica), imunofenotipska (izražaj antigena na staničnoj površini) te citogenetska i molekularna obilježja (kromosomske abnormalnosti i genske mutacije).
Morfološka obilježja. Maligne leukemijske stanice kod bolesnika su obično veličine 12 do 20 nm (u promjeru), a karakteriziraju ih višestruke jezgrice, citoplazma koja sadrži azurofilne granule te patognomonične fuziformne citoplazmatske inkluzije zvane Auerovi štapići. Prema morfološkim obilježjima i sličnostima s određenim stanicama mijeloidne loze, akutne mijeloične leukemije dijelimo u osam podtipova (tzv. FAB klasifikacija, engl. French-American-British morphology system), kako je navedeno u Tablici 6.24.
|
Tablica 6.24. FAB klasifikacija AML-a
|
|
Oblik
|
Naziv
|
Imunofenotip
|
|
M0
|
Leukemija s minimalnom diferencijacijom
|
CD34, CD17, HLA-DR
|
|
M1
|
Mijeloična leukemija bez sazrijevanja
|
CD34, CD17, HLA-DR
|
|
M2
|
Mijeloična leukemija sa sazrijevanjem
|
CD34, CD17, HLA-DR
|
|
M3
|
Hipergranularna promijelocitna leukemija
|
CD13, CD33, CD14, CD15, CD11b
|
|
M4
|
Mijelomonocitna leukemija
|
CD13, CD33, CD14, CD15, CD11b
|
|
M5
|
Monocitna leukemija
|
CD13, CD33, CD14, CD15, CD11b
|
|
M6
|
Eritroleukemija
|
CD13, CD33, CD36, CD71
|
|
M7
|
Megakarioblastna leukemija
|
CD13, CD33, CD41, CD61
|
Imunofenotipska obilježja. Većina leukemijskih stanica bolesnika s AML-om na svojoj površini eksprimira klasične antigene kakvi se viđaju na površini stanica mijeloidne loze zdravih ljudi. Određivanje vrste antigena na staničnoj površini (protočna citometrija) često pomaže pri diferencijaciji porijekla malignoga klona te može imati određeni dijagnostički i prognostički značaj. Slabije diferencirani oblici AML-a (M0-M2) na svojoj površini sadrže antigene CD34, CD117 te HLA-DR, dok jasnije diferencirani oblici (M3-M7) tipično na svojoj površini sadrže antigene CD13 i CD33. AML monocitnoga porijekla (M3-M5) često sadrže i CD14, CD15 i CD11b antigene, eritroidnoga porijekla (M6) CD36 i CD71 antigen, a megakariocitnoga porijekla (M7) CD41 i CD61 antigen.
Citogenetska i molekularna obilježja. Određeni oblici akutne mijeloične leukemije povezani su s karakterističnim kromosomskim poremećajima, bilo da se radi o poremećajima njihova broja, bilo strukture. Najčešći kromosomski poremećaji kod takvih bolesnika su kromosomske translokacije kao što su t(8;21), t(15;17), t(11q23), t (9;11) ili t(11;19), kromosomske inverzije kao što je inv(16) te druge abnormalnosti, kao što su delecija dijela ili cijeloga 7. ili 9. kromosoma, trisomije 8., 11. ili 21. i sl. Poznavanje navedenih kromosomskih poremećaja korisno je u predviđanju terapijskoga odgovora i prognoze, a neki imaju i snažnu dijagnostičku ulogu, kao npr. translokacija t(15;17) kod bolesnika s akutnom promijelocitnom leukemijom. Navedene kromosomske promjene rezultiraju poremećajem ekspresije gena uključenih u proces stanične proliferacije i diferencijacije mijeloidnih stanica, što za posljedicu ima njihovu malignu preobrazbu. Neki od najčešćih patološki eksprimiranih gena kod ovih bolesnika su RUNX1-RUNX1T1, CBFB-MYh11, PML-RAR alfa, MLLT3-MLL, DEK-NUP214 i dr.
SZO klasifikacija AML-a. Uzimajući u obzir sva navedena obilježja akutnih mijeloičnih leukemija, Svjetska zdravstvena organizacija donijela je univerzalnu klasifikaciju akutnih mijeloičnih leukemija koja se danas i koristi u svakodnevnoj kliničkoj praksi (Tablica 6.25.).
|
Tablica 6.25. Klasifikacija AML-a prema Svjetskoj zdravstvenoj organizaciji
|
|
AML s povratnim genetskim promjenama
AML s t(8;21)(q22,q22); RUNX1-RUNX1T1
AML s inv(16)(p13.1q22); CBFB-MYh11
APL s t(15;17)(q22;q12); PML-RARA
AML s t(9;11)(p22;q23); MLLT3-MLL
AML s t(6;9)(p23;q34); DEK-NUP214
AML s inv(3)(q21q26.2) ili t(3;3)(q21;q26.2); RPN-EVI1
AML (megakarioblastna) s t(1;22)(p13;q13); RBM15-MKL1
Privremeni podtip: AML s mutacijom NPM1
Privremeni podtip: AML s mutacijom CEBPA
|
|
AML sa znakovima mijelodisplazije
|
|
AML nakon terapije
|
|
AML koje nisu svrstane drugamo
AML s minimalnom diferencijacijom
AML bez sazrijevanja
AML sa sazrijevanjem
Akutna mijelomonocitna leukemija
Akutna monoblastna/monocitna leukemija
Akutna eritroidna leukemija
Akutna megakarioblastična leukemija
|
|
Mijeloidni sarkom
|
|
Mijeloidne proliferacije pri Downovu sindromu
Prolazni poremećaj mijelopoeze
Mijeloidna leukemija pri Downovom sindromu
|
|
Tumor blastičnih plazmacitoidnih dendritičkih stanica
|
Klinička slika. Početni simptomi i znakovi akutnih mijeloičnih leukemija su nespecifični te uključuju brzo umaranje, slabost, gubitak teka te pad na tjelesnoj težini, a postupno se razvijaju i znakovi neefektivne mijelopoeze: eritrocitopenija i anemični sindrom, trombocitopenija i hemoragijska dijateza (najčešće u obliku potkožnih krvarenja – petehije, ekhimoze, hematomi) te učestale infekcije kao posljedica nefunkcionalnih leukocita (osobito kod pada vrijednosti neutrofila ispod 0,5 x 109/L). Hepatosplenomegalija i periferna limfadenopatija nastaju kao posljedica infiltracije leukemijskim stanicama koje pokazuju sklonost prema tim tkivima, iako mogu infiltrirati svaki organ ili tkivo. Kod bolesnika s izraženom leukocitozom može se razviti i hiperleukocitarni sindrom, odnosno sindrom leukostaze primarno obilježen poremećajem mikrocirkulacije s posljedičnom sklonosti krvarenju i/ili formiranju leukocitnih tromba s ishemijskim oštećenjima tkiva i organa (mozak, pluća, bubrezi, srce), a mogu se javiti i znaci staničnoga hipermetabolizma (hiperurikemija i njezine posljedice, hiperosmolarnost, hiperkalijemija, metabolička acidoza, dehidracija i sl.). Određeni oblici akutnih mijeloičnih leukemija mogu biti povezani i sa specifičnim kliničkim manifestacijama, kao što je hipertrofija gingive i infiltracija kože kod bolesnika s akutnom mijelomonocitnom i monocitnom leukemijom (M4, M5) ili sklonost razvoju diseminirane intravaskularne koagulopatije kod bolesnika s akutnom promijelocitnom leukemijom (M3). Brzina razvoja simptoma i progresija bolesti u prvom redu ovisi o tipu akutne leukemije te prisutnosti povoljnih ili nepovoljnih prognostičkih čimbenika (kasnije opisano).
Dijagnoza akutne mijeloične leukemije počiva na laboratorijskim nalazima periferne krvi te citološkoj punkciji i biopsiji koštane srži. Karakteristično u osnovnim laboratorijskim nalazima nalazimo različito izraženu leukocitozu (od 15 do > 100 x 109/L leukocita) uz nalaz više od 20 % nezrelih oblika stanica (mijeloblasti) u diferencijalnoj krvnoj slici te različito izraženu anemiju, granulocitopeniju (neutropenija) i/ili trombocitopeniju. Osim toga, za potvrdu bolesti nužna je i citološka punkcija i biopsija koštane srži koja je od presudne važnosti u diferenciranju vrste akutne mijeloične leukemije. Ona omogućava morfološki pregled koštane srži, a dodatno se provodi još i imunofenotipizacija, klasična citogenetika (G-pruganje) te molekularna dijagnostika (RT-PCR). Morfološki gledano, koštana srž bolesnika je hipercelularna te sadrži 20 do 100 % blasta (mijeloblasti) koji potiskuju normalno hematopoetsko tkivo. Također, ovisno o vrsti leukemije, dodatno se može naći različito izražena mijelofibroza (osobito u AML M7) ili nekroza. Imunofenotipizacija uzorka koštane srži (protočna citometrija) omogućava određivanje antigenske ekspresije leukemijskih stanica što može ukazivati na stanično porijeklo, a standardno se izvodi i klasična citogenetika koja može ukazati na karakteristične kromosomske aberacije te molekularna dijagnostika kojom se ispituju postojanje NPM1, CEBPA i FLT3 mutacije. Ostali laboratorijski nalazi ovise o težini bolesti (hiperurikemija, povišene vrijednosti laktat dehidrogenaze, koagulogram i sl.). Ovisno o kliničkoj sumnji na zahvaćenost određenih organa, potrebno je provesti i ciljane slikovne radiološke metode (rendgenogram ili kompjuterizirana tomografija prsnog koša, ultrazvuk abdomena, lumbalna punkcija i sl.).
Prognostički faktori. Poznavanje definiranih prognostičkih faktora kod bolesnika s AML-om ključno je za donošenje odluke o načinu (modalitetu) liječenja, s obzirom na to da se ona razlikuje u odnosu na prisutnost/odsutnost određenih faktora. Prognostički faktori mogu ukazivati na uspješnost postizanja remisije, kao i na trajanje terapijskoga odgovora (trajanje remisije). U načelu se bolesnici s nepovoljnijim prognostičkim faktorima moraju liječiti agresivnijim protokolima u odnosu na bolesnike s povoljnijim prognostičkim faktorima. Prognostički faktori bolesnika s AML-om prikazani su u Tablici 6.26.
|
Tablica 6.26. Prognostički faktori AML-a
|
|
Povoljni prognostički faktori
|
Nepovoljni prognostički faktori
|
|
Dob < 50 godina
Karnofsky skor > 60 %
Novonastala (de novo) leukemija
CD34 negativni fenotip
t(15;17), t(8;21), inv(16) ili varijanta t(16;16)
Uredni kromosomi 5 i 7
Prisutnost NPM1 mutacije
|
Dob > 60 godina
Karnofsky skor < 60 %
Leukemija nastala iz MDS-a ili nakon liječenja
CD34 pozitivan fenotip
inv(3) ili t(3;3), t(6;9)
Promjene kromosoma 5 ili 7 (delecija, trisomija)
Prisutnost FLT3 mutacije
|
Liječenje. Osnovu liječenja akutne mijeloične leukemije čine kemoterapija i transplantacija koštane srži. S obzirom na to da se radi o brzo progresivnoj bolesti, liječenje bi trebalo započeti što ranije od postavljanja dijagnoze, najbolje već u prva 72 sata. Samo liječenje provodi se kroz dvije faze: indukcijsko i konsolidacijsko liječenje. Indukcijsko se liječenje naziva još i uvodna terapija, a podrazumijeva primjenu citostatika s ciljem uvođenja bolesnika u kompletnu remisiju, dok konsolidacijska terapija za cilj ima održati kompletnu remisiju bolesti, odnosno postići izlječenje. Kompletna remisija podrazumijeva smanjenje blasta na manje od 5 % u koštanoj srži, te 0 u perifernoj krvi uz rezoluciju citopenije i poboljšanje kliničkoga stanja. Protokoli liječenja bolesnika s akutnom mijeloičnom leukemijom razlikuju se ovisno o vrsti leukemije, dobi bolesnika te prisutnosti prethodno navedenih prognostičkih faktora (povoljni, nepovoljni).
Indukcija. Uvodna ili indukcijska terapija za cilj ima postizanje kompletne remisije bolesti. Način i protokol liječenja u prvom redu ovise o dobi i kondicijskom stanju bolesnika. Klasičnu indukcijsku terapiju za većinu bolesnika čini kombinirana kemoterapija (antraciklin - daunorubicin ili idarubicin plus citarabin) kojom se postiže kompletna remisija kod 80 – 90 % osoba do 60 godina starosti te 50 – 60 % osoba u dobi iznad 60 godina. Kod starijih osoba, kao i mlađih s lošijim kondicijskim stanjem, koji nisu kandidati za ovu tradicionalnu kemoterapiju, primjenjuju se manje agresivni lijekovi kao što su 5-azacitadin, decitabin i klofarabin s relativno prihvatljivim rezultatima. Primjena bcl2 inhibitora (venetoklaks) u indukcijskom liječenju pokazuje bolje ishode u postizanju kompletne remisije, ali je još uvijek u fazi kliničkih ispitivanja. Također, određene studije pokazale su dodatnu korist od primjene monoklonalnoga protutijela anti-CD33 (gemtuzumab, ozogamicin) u indukcijskom i konsolidacijskom liječenju s ciljem postizanja dugoročnije remisije bolesti.
Konsolidacija. Konsolidacijska terapija provodi se nakon indukcijskoga liječenja s ciljem eradikacije preostalih leukemijskih stanica i dugotrajnijega održavanja postignute remisije. Ona se sastoji od intenzivirane standardne kemoterapije s ili bez alogene transplantacije koštane srži. Način provođenja indukcijskoga liječenja je individualan i zasniva se na citogenetskom i molekularnom nalazu (Tablica 6.27.). Kod bolesnika s povoljnom citogenetikom u indukcijskom liječenju primjenjuje se samo standardna kemoterapija (tri do četiri ciklusa inicijalne kemoterapije) bez alogene transplantacije, budući da ona ne povećava znatno postotak održavanja remisije, a nosi znatan rizik od komplikacija. Kod bolesnika s citogenetikom srednjega rizika i nepovoljnom citogenetikom uz standardnu se kemoterapiju preporuča i alogena transplantacija koštane srži, budući da se njome znatno povećava period kompletne remisije (autologna transplantacija predstavlja alternativnu opciju za bolesnike kojima se ne može naći podudarni donor koštane srži, a kod starijih bolesnika može se pristupiti alogenoj transplantaciji s pripremom smanjena intenziteta). Takvim se pristupom dugotrajna kompletna remisija postiže kod oko 80 % bolesnika mlađih od 60 godina. Osim standarne kemoterapije, kod pacijenata s FLT3 mutacijom u liječenju se koristi i midostarurin FTL3 inhibitor.
|
Tablica 6.27. Prognostički značaj citogenetskoga i molekularnog nalaza bolesnika s AML-om
|
|
Povoljna citogenetika
|
Citogenetika srednjeg rizika
|
Nepovoljna citogentika
|
|
t(8;21)(q22;q22)
Mutacija RUNX1-RUNX1T1
inv(16)(p13.1q22)
t(16;16)(p13.1;q22)
Mutacija CBFB-MYH11
Mutacija NPM1 bez FLT3
|
Mutacija NPM1 i FLT3
Mutacija FLT3 bez NPM1
t(9;11)(p22;q23)
Mutacija MLL3-MLL
|
inv(3)(q21q26.2)
t(3;3)(q21;q26.2)
Mutacija RPN1-EVI1
t(6;9)(p23;q34)
Mutacija DEK-NUP214
del(5q)
|
Liječenje relapsa AML. Povrat bolesti praćen je lošijim ishodima u pogledu ponovnoga postizanja remisije. I kod tih se bolesnika provodi indukcijsko i konsolidacijsko kemoterapijsko liječenje, a dugotrajno preživljenje rijetko se postiže bez alogene transplantacije koštane srži te ju treba obaviti kod svih bolesnika. Kod te skupine bolesnika u obzir dolazi i primjena ciljanih lijekova, kao što je to enasidenib kod bolesnika s IDH1/IDH2 mutacijom.
Liječenje akutne promjelocitne leukemije. Liječenje ove vrste leukemije izdvaja se od načina liječenja ostalih vrsta leukemija. Kako je već opisano, akutnu promijelocitnu leukemiju karakterizira translokacija t(15;17) i izražena sklonost razvoju koagulopatije i krvarenja. Osnovu liječenja čini primjena transretinoične kiseline (ATRA) koja dovodi do brze diferencijacije promijelocita i smanjenja rizika od krvarenja. Uz ATRA-u primjenjuje se i kemoterapija temeljena na antraciklinima u indukcijskom liječenju uz dva ili tri ciklusa konsolidacijske kemoterapije, također temeljene na antraciklinima uz ATRA-u. Terapija održavanja (6-merkaptopurin, meotreksat, ATRA) ima povoljan učinak kod bolesnika liječenih indukcijom i konsolidacijom. Stopa remisije uz taj način liječenja iznosi oko 90 % bolesnika mlađe životne dobi, a u preko 70 % slučajeva ona dovodi i do izlječenja. Alogena transplantacija koštane srži indicirana je ukoliko se primjenom ATRA-e ne postiže zadovoljavajuća kontrola bolesti ili u sklopu konsolidacijskoga liječenja nakon relapsa bolesti. U rezistentnom ili relapsirajućem obliku bolesti u obzir dolazi i primjena arsenova trioksida koji ima sposobnost izazivanja apoptoze promijelocita.
Potporno liječenje obuhvaća podržavajuće mjere koje se provode zajedno s aktivnim liječenjem: 1) korekcija anemije (primjena transfuzije koncentriranih eritrocita s ciljem održavanja vrijednosti hemoglobina između 80 i 100 g/L), 2) prevencija krvarenja (primjena transfuzije trombocita s ciljem održavanja broja trombocita 5 – 10 x 109/L ukoliko nema krvarenja ili > 50 x 109/L u slučaju krvarenja ili podvrgavanja određenim invazivnim postupcima), 3) korekcija koagulacijskih abnormalnosti (održavanje vrijednosti INR-a i fibrinogena primjenom svježe smrznute plazme ili koncentrata fibrinogena), 4) profilakstička i terapijska kontrola infekcija te 5) korekcija metaboličkih poremećaja (npr. alopurinol kod hiperurikemije)..
Prognoza bolesnika s AML-om ponajprije ovisi o dobi, kliničkom stanju i citogenetskom, odnosno molekularnom statusu. Ukupno gledano, oko 80 % bolesnika mlađih od 60 godina postigne kompletnu remisiju, dok je taj postotak niži kod starijih osoba i iznosi oko 50 %. Kod osoba mlađe životne dobi veći su izgledi za potpuno izlječenje (trajna remisija), dok su kod starijih znatno niži (svega 10 %). Najbolje dugoročne ishode imaju bolesnici liječeni alogenom transplantacijom koštane srži.
AKUTNA LIMFOBLASTIČNA LEUKEMIJA
Definicija. Akutna limfoblastična leukemija (ALL) predstavlja akutnu leukemiju koja nastaje uslijed prekomjernoga stvaranja i nakupljanja nezrelih limfocita (limfoblasta) u koštanoj srži, a potom i perifernoj krvi i drugim organima. Prema podjeli Svjetske zdravstvene organizacije, ta se leukemija zajedno s limfomima ubraja u jedinstvenu skupinu limfoproliferativnih bolesti predstavljajući zapravo istu bolest s dvije kliničke prezentacije, a to su leukemija ili limfom. O akutnoj limfoblastičnoj leukemiji govori se kada se u koštanoj srži nalazi više od 20 % limfoblasta, bez obzira na prisutnost nodalne ili ekstranodalne limfomske mase, dok nalaz manji od 20 % limfoblasta u koštanoj srži uz dokazanu limfomsku masu označava limfom..
Epidemiologija. ALL predstavlja najčešću malignu tumorsku bolest djece (s najčešćom pojavnosti između 2. i 10. godine života), dok se kod odraslih javlja nešto rjeđe (2 slučaja na 100 000 stanovnika godišnje), i to obično u drugom i trećem desetljeću života.
Etiopatogeneza. Kao i kod akutne mijeloične leukemije, točan uzrok i mehanizam nastanka bolesti nisu poznati. U razvoju bolesti određenu ulogu mogu imati genetska predispozicija, izloženost ionizirajućem zračenju, infekcije onkogenim virusima (humani T-leukemijski virus, Epstein-Barrov virus) te različite kemikalije i lijekovi. ALL je klonalna bolest koja započinje patološkom proliferacijom i nakupljanjem limfoblasta u koštanoj srži, a potom se širi u perifernu krvi i druge organe. ALL za razliku od ALM ima veću sklonost infiltraciji limfatičnih tkiva, solidnih organa i središnjega živčanog sustava.
Patogenetska obilježja. Akutna limfoblastična leukemija, kao i akutna mijeloična leukemija, pokazuje određene karakteristike u morfologiji stanica, imunofenotipu, citogenetici i molekularnim obilježjima.
Morfološka obilježja. Općenito gledano, leukemijske stanice bolesnika s ALL-om manje su u odnosu na stanice bolesnika s AML-om. Ovisno o njihovim karakteristikama, ALL se dijeli u tri podskupine prema FAB klasifikaciji: L1, L2 i L3. ALL-L1 tipa čini 75 % slučajeva, a obilježena je malim i okruglim limfoblastima s oskudnom citoplazmom u kojoj se nadziru pokoje, rijetke granulacije. ALL-L2 čini oko 20 % slučajeva, a karakteriziraju je veliki i pleomorfni limfoblasti s istaknutom jezgricom, dok je ALL-L3 najrjeđi oblik na koji otpada oko 5 % slučajeva, a karakterizirana je velikim limfoblastima s jasno istaknutom jezgrom i bazofilnom citoplazmom. Morfološko razlikovanje T ili B-staničnoga porijekla limfoblasta nije moguće, a često je otežana i diferencijacija prema mijeloblastima, zbog čega tu presudnu ulogu ima imunofenotipizacija.
Imunofenotipska obilježja. Ovisno o antigenskome izražaju na staničnoj površini razlikuju se B-limfoblastična leukemija (B-ALL) i T-limfoblastična leukemija (T-ALL) koje se dodatno dijele u nekoliko podskupina ovisno o vrsti antigena koje eksprimiraju (Tablica 6.28.).
|
Tablica 6.28. Imunofenotipska podjela akutne limfoblastične leukemije
|
|
B-limfoblastična leukemija (B-ALL)
|
|
Rana prekursorska B-ALL (pro-B-ALL)
|
CD19, CD22, CD79a
|
|
Common ALL
|
CD10+ (zajednički ili common antigen, CALLA)
|
|
Kasna B-ALL (pre-B-ALL)
|
CD20
|
|
T-limfoblastična leukemija (T-ALL)
|
|
Rana prekursorska T-ALL (pro-T-ALL)
|
CD7+, CD2-, CD4-, CD8-
|
|
Pre-T-ALL
|
CD2+, CD7+, CD4-, CD8-
|
|
Kortikalna T-ALL
|
CD1+, CD7+, CD2+, CD5+CD4+, CD8+
|
|
Zrela T-ALL
|
CD3+, CD2+, CD7+
|
Citogenetska i molekularna obilježja. Kao i ostale, i ova je vrsta leukemije povezana sa specifičnim kromosomskim i genetskim obilježjima. Neki od tih poremećaja povezani su boljim, a neki s lošijim ishodima. Najvažniji i najčešći povoljni citogenetski poremećaji uključuju translokaciju t(12,21)(TEL-AML1) te hiperdiploidiju (više od 50 kromosoma), dok nepovoljni citogenetski poremećaji uključuju translokaciju t(9;22) poznatu kao Philadelphia kromosom, translokaciju t(4;11)(MLL) te hipodiploidiju (manje od 44 kromosoma). Ti se poremećaji dokazuju klasičnim metodama kao što je G-pruganje, FISH metoda ili RT-PCR.
SZO klasifikacija ALL. Uzimajući u obzir navedena imunofenotipska, citogenetska i molekularna obilježja leukemijskih stanica razlikuje se nekoliko oblika akutne B i T-limfoblastične leukemije/limfoma (Tablica 6.29.).
|
Tablica 6.29. SZO klasifikacija akutne limfoblastične leukemije/limfoma
|
|
B-akutna limfoblastična leukemija/limfom nespecificirani
|
|
B-akutna limfoblastična leukemija/limfom s povratnim genetskim promjenama
B-ALL/LBL s t(9;22)(q34;q11.1); BCR-ABL1
B-ALL/LBL s t(v;11q23); MLL preustroj
B-ALL/LBL s t(12;21)(p13;q22); TEL-AML1 (ETV6-RUNX1)
B-ALL/LBL s t(5;14)(q31;q32); IL3-IGH
B-ALL/LBL s t(1;19)(q23;p13.3) TCF3-PBX1
|
|
B-akutna limfoblastična leukemija/limfom s hiperdiploidijom
|
|
B-akutna limfoblastična leukemija/limfom s hipodiploidijom
|
|
T-akutna limoblastična leukemija/limfom
|
Klinička slika. Kliničke manifestacije ALL-a mogu biti posljedica infiltracije koštane srži i njezine posljedične disfunkcije, infiltracije drugih organa i tkiva te staničnoga hipermetabolizma. Kao posljedica slabosti koštane srži, kod bolesnika se razvijaju anemija s pridruženim simptomima, hemoragijska dijateza (trombocitopenija) te učestale infekcije (granulocitopenija). Za razliku od AML-a, kod oboljelih od ALL-a znatno se češće pojavljuje infiltracija drugih organa i tkiva leukemijskim stanicama, osobito limfatičnoga tkiva, kostiju i kože, što se manifestira odgovarajućim simptomima i znakovima, a česta je i infiltracija središnjega živčanog sustava. Karakteristična je i pojava tumorske mase u prednjem medijastinumu u bolesnika s T-ALL-om. Ona može biti praćena nastankom pleuralnoga izljeva, sindroma gornje šuplje vene, kao i drugih kompresivnih sindroma. Uz to, prisutni su i klasični znaci staničnoga hipermetabolizma kao što su umor, malaksalost, gubitak teka i tjelesne težine, povišena tjelesna temperatura i sl.
Dijagnoza ALL-a postavlja se na osnovi laboratorijskih nalaza (kompletna i diferencijalna krvna slika), citomorfološkoga nalaza uzorka koštane srži s imunofenotipizacijom leukemijskih stanica te punkcijom/biopsijom drugih zahvaćenih tkiva i organa. U laboratorijskim nalazima povećan je broj leukocita s udjelom blasta većim od 20 % u diferencijalnoj krvnoj slici. Ovisno o težini bolesti pojavljuje se i različito izražena anemija, trombocitopenija i neutropenija. Za potvrdu dijagnoze ALL-a nužna je punkcija ili biopsija koštane srži u kojoj se nalazi više od 20 % blasta (ako se radi o udjelu manjem od 20 %, govori se o limfomu), a imunofenotipizacijom se dokazuje prisutnost antigena B-stanične linije (CD10, CD19) ili T-stanične linije (CD3, CD4, CD8). Također, gotovo sve leukemijske stanice bolesnika s ALL-om eksprimiraju na svojoj površini DNA polimerazu TdT (engl. terminal deoxynucleotidyl transferase). Citogenetske (G-pruganje) i molekularne metode (RT-PCR) služe za određivanje citogenetskih i molekularnih obilježja što ima ulogu u klasifikaciji i predviđanju prognoze bolesnika. Dijagnostički se postupak nadopunjava i drugim dijagnostičkim metodama kojima se otkriva raširenost bolesti, ovisno o simptomima i indikacijama (rendgenogram ili CT prsnog koša, biopsija ili punkcija limfnoga čvora, lumbalna punkcija i sl.).
Prognostički čimbenici. Poznavanje prognostičkih čimbenika, osim za predviđanju ishoda bolesti, bitnu ulogu ima i u određivanju terapijskoga pristupa. Rizični čimbenici koji određuju ishod bolesti i oblik liječenja su dob, broj leukocita u perifernoj krvi, imunofenotip, citogenetske promjene, početni odgovor na terapiju te ekstramedularnu proširenost bolesti (Tablica 6.30.).
|
Tablica 6.30. Prognostički čimbenici kod bolesnika s akutnom limfoblastičnom leukemijom
|
|
Rizični čimbenici
|
Pokazatelji povoljnoga ishoda
|
Pokazatelji nepovoljnoga ishoda
|
|
Dob
|
Mlađa životna dob (< 40 godina)
|
Starija životna dob (> 40 godina)
|
|
Broj leukocita
|
< 50 x 109/L (B-ALL); < 100 x 109/L (T-ALL)
|
> 50 x 109/L (B-ALL); > 100 x 109/L (T-ALL)
|
|
Imunofenotip
|
CD10+
|
Pro-B-ALL
|
|
Citogenetske promjene
|
t(12;21), hiperdiploidija
|
t(9;22), t(4;11), hipodiploidija
|
|
Početni odgovor na terapiju
|
Rani nestanak blasta (< 4 tjedna)
|
Bez remisije nakon 4 tjedna
|
|
Ekstramedularna proširenost
|
Bez ekstramedularne proširenosti
|
Prisutna ekstramedularna proširenost
|
Liječenje. Kao i kod AML-a, liječenje se sastoji od indukcije (uvodna terapija) kojom se nastoji postići remisija, a potom od konsolidacije kojom se nastoji održati postignuta remisija, što uključuje kemoterapiju i/ili transplantaciju koštane srži. Remisija se definira stanjem u kojemu se blasti ne mogu detektirati u perifernoj krvi i koštanoj srži uz oporavljenu mijelopoezu (oporavak krvne slike). Za razliku od bolesnika s AML-om, kod ovih je bolesnika indicirana profilaksa središnjega živčanog sustava zbog sklonosti prema njegovu zahvaćanju, a obično se to provodi intratekalnom primjenom kemoterapeutika.
Indukcija. Uvodnu terapiju ALL-a kod odraslih bolesnika čini kombinirana kemoterapija koja uključuje primjenu danorubicina, vinkristina, asparaginaze te glukokortikoida, a bolesnicima koji imaju Philadelphia kromosom dodaje se i inhibitor tirozin kinaze, kao što je imatinib ili dasatinib. Takvim se režimom kompletna remisija postiže se kod oko 90 % bolesnika. Navedeni kemoterapeutici imaju selektivniji učinak i ne moraju dovesti do značajne mijelosupresije i aplazije koštane srži.
Konsolidacija. Nastavak liječenja nakon postignute remisije ovisi o dobi bolesnika i prognostičkim čimbenicima. Kod mlađih bolesnika s povoljnim prognostičkim čimbenicima liječenje se temelji na nastavku primjene kemoterapije pri čemu se obično koriste kemoterapeutici različiti od onih kojima je postignuta remisija (metotreksat, ciklofosfamid, citarabin), dok se kod bolesnika s nepovoljnim prognostičkim čimbenicima, kao i onima koji slabije reagiraju na primijenjeno liječenje, preporučuje i alogena transplantacija koštane srži. Nakon provedenoga konsolidacijskog liječenja preporučeno je nastaviti terapiju održavanja koja se sastoji od primjene kemoterapeutika u malim dozama na dnevnoj ili tjednoj razini (npr. 6-merakaptopurin svakodnevno plus metotreksat na tjednoj ili dvotjednoj bazi) kroz razdoblje od dvije do tri godine.
Profilaksa središnjega živčanog sustava. Većina kemoterapeutika koja se primjenjuje u standardnoj dozi ne postiže odgovarajuće terapijske koncentracije u središnjem živčanom sustavu te se kod više od 35 % bolesnika može očekivati pojava recidiva bolesti u središnjemu živčanom sustavu. Terapijske koncentracije u središnjem živčanom sustavu mogu se očekivati samo kod bolesnika kod kojih se primjenjuju visoke doze metotreksata i citarabina, dok je kod ostalih indicirana profilaksa u smislu intratekalne primjene metotreksata s ili bez zračenja neurokranija. Uz opisanu profilaksu stopa recidiva bolesti u središnjemu živčanom sustavu manja je od 10 %.
Relaps i refrakterni ALL. Liječenje relapsa obično se provodi istim kemoterapeuticima kojima je postignuta i prva remisija, a nakon postizanja ponovne remisije (što se obično dogodi u 50 do 70 % slučajeva) indicirano je liječenje alogenom transplantacijom koštane srži. Također, u liječenju relapsa kao i refrakternih oblika ALL-a na raspolaganju su i noviji lijekovi, kao što su nelarabin (inhibitor DNA sinteze), blinatumomab za B-ALL CD19+ (bispecifični aktivator T-limfocita koji stvara citolitičke sinapse zmeđu T-limfocita i leukemijskih stanica B staničnog porijekla vežući se za protein CD19) te inotuzumab ozogamicin za B-ALL CD22+ (konjugat monoklonskoga protutijela usmjerenoga protiv CD22 putem kojega lijek i dospijeva u tumorsku stanicu, a na njega je vezan N-acetil-gama-kalikeamicindimetilhidrazid koji ima citotoksičan učinak).
Prognoza. Iako ALL ima izvrsnu prognozu kod djece, kod odraslih je bolesnika ona nešto lošija. S povećanjem dobi povećava se i učestalost pojave Philadelphia kromosoma koji se smatra lošim prognostičkim čimbenikom (kod djece je prisutan u oko 5 % slučajeva, a kod odraslih u više od 25 % slučajeva). Kompletnu remisiju postigne oko 80 % bolesnika, dok se izlječenje očekuje kod oko 60 % mlađih bolesnika i bolesnika s povoljnom citogenetikom, a taj je postotak znatno niži kod starijih bolesnika i bolesnika s nepovoljnom citogenetikom - iznosi svega 20 do 40 %.
Najznačajniji oblici kroničnih leukemija su kronična mijeloična leukemija, kronična limfocitna leukemija i leukemija vlasastih stanica. Kronična mijeloična leukemija opisana je u poglavlju „Kronične mijeloproliferativne bolesti“.
KRONIČNA LIMFOCITNA LEUKEMIJA
Definicija. Kronična limfocitna leukemija (KLL) najčešći je oblik leukemije koji se susreće u kliničkoj praksi, a karakterizirana je pojačanim stvaranjem i nakupljanjem zrelih limfocita u koštanoj srži i perifernoj krvi. U više od 95 % slučajeva radi se o B-limfocitima, dok se u ostatku (< 5 %) radi o T-limfocitima ili NK-stanicama. Prema klasifikaciji Svjetske zdravstvene organizacije, KLL se zajedno s limfomom malih stanica ubraja u neoplazme zrelih B-stanica, a njihova je osnovna razlika u mjestu nastanka bolesti (KLL nastaje u koštanoj srži i krvi, dok limfom malih stanica nastaje u limfnom tkivu). U nastavku teksta bit će opisan samo leukemijski oblik bolesti, dok je limfom malih stanica opisan u poglavlju o ne-Hodgkinovim limfomima.
Epidemiologija. Kako je već navedeno, u zapadnim zemljama KLL predstavlja najčešći oblik leukemije s prosječnom incidencijom od 3 do 4 novootkrivena bolesnika na 100 000 stanovnika. Bolest se javlja gotovo isključivo kod odraslih osoba (pojava prije tridesete godine izuzetno je rijetka), a većina oboljelih osobe su starije od 60 godina. Također, bolest se češće pojavljuje kod muškaraca u odnosu na žene, i to u omjeru od 2:1.
Etiopatogeneza. KLL nastaje kao posljedica klonalne ekspanzije zrelih B-limfocita (rijetko T-limfocita T ili NK-stanica), a točan uzrok i mehanizam te ekspanzije ostaju nepoznati. Osim nasljeđa kao mogućega rizičnog čimbenika (osobe koje u prvom koljenu imaju rođaka s KLL-om imaju dva to tri puta veći rizik od obolijevanja, i to u mlađoj dobi u odnosu na opću populaciju), kao mogući etiološki faktori spominju se i neki herbicidi i pesticidi jer je zamijećeno da poljoprivrednici češće obolijevaju od KLL-a u odnosu na druge profesije i opću populaciju. Povezanost ionizirajućega zračenja i virusnih infekcija s pojavnosti KLL-a nije dokazana. Kao i kod ostalih hematoloških oboljenja, i kod oboljelih od KLL-a prisutne su određene mutacije i kromosomske promjene koje predisponiraju razvoj KLL-a. Najčešće genetske mutacije povezane s razvojem KLL-a su TP53 (15 %), SF3B1 (15 %), MYD88 (10 %), ATM (9 %) te NOTCH1 (4 %), dok se od kromosomskih anomalija najčešće susreće delecija dugog kraka 13. kromosoma (13q), delecija dugog kraka 11. kromosoma (11q), trisomija 12. kromosoma te delecija kratkog kraka 17. kromosoma (17p). Prisutnost delecije 17p i 11q povezana je s lošijim ishodima, dok je delecija 13q povezana s boljim kliničkim ishodima.
Citomorfološka obilježja. Tumorske stanice KLL-a homogenoga su izgleda, a posjeduju morfologiju normalnih, zrelih limfocita. Površinu stanica karakterizira prisutnost monoklonalnih površinskih imunoglobulina (IgM i IgD), te pan-B staničnih biljega (CD5, CD19, CD20, CD23, CD24), odnosno T-staničnih biljega ako se radi o KLL-u T-staničnoga porijekla. Osim toga, na površini većine stanica nalazi se i Ia antigen te receptor za Fc fragment imunoglobulina G (IgG).
Patofiziologija. U KLL-u maligna transformacija i umnažanje zrelih limfocita nastupaju u koštanoj srži, a potom se oni nakupljaju u perifernoj krvi, a u krajnjoj liniji mogu infiltrirati i različite organe i tkiva kao što su limfni čvorovi, slezena, koža, organi probavnoga i dišnog sustava i dr. Uslijed umnažanja leukemijskoga klona u koštanoj srži, dolazi do potiskivanja stanica mijelopoeze što rezultira anemijom i trombocitopenijom. Također, nastali B-limfociti obično imaju oslabljenu funkciju, zbog čega dolazi do hipogamaglobulinemije i sklonosti infekcijama.
Klinička slika i tijek bolesti. Kod većine je bolesnika KLL asimptomatska bolest i dijagnosticira se slučajnim nalazom povećanoga broja limfocita u perifernoj krvi. Kod većine ima benigan, sporo-progresivan tijek te se često opisuje kao leukemija s kojom se živi, a od koje se ne umire. Nespecifični simptomi poput umora, malaksalosti, gubitka teka i tjelesne težine mogu biti prisutni, ali nisu specifični. S napredovanjem bolesti pojavljuju se manifestacije mijelosupresije i imunodeficijencije, zbog čega su bolesnici blijedi, skloni krvarenju te imaju učestale infekcije, osobito one uzrokovane gljivama i gram-negativnim bakterijama. Kao posljedicu leukemijske infiltracije bolesnici mogu imati generaliziranu limfadenopatiju te hepatosplenomegaliju (u trenu postavljanja dijagnoze oko 80 % bolesnika ima difuznu limfadenopatiju, a oko 50 % bolesnika ima umjerenu hepatosplenomegaliju). Infiltracija središnjega živčanog sustava je rijetka te neurološki simptomi trebaju prije upućivati na oportunističku infekciju (kriptokokoza, listerioza), nego na diseminaciju bolesti. 5 do 10% bolesnika s KLL-om razvije autoimunu hemolitičku anemija ili autoimunu trombocitopeniju, dok se u oko 5 % slučajeva ona transformira u velikostanični limfom (Richterov sindrom).
Dijagnoza se postavlja na temelju laboratorijskih nalaza te biopsije i/ili citološke punkcije koštane srži uz imunofenotipizaciju te citogenetsku i molekularnu dijagnostiku.
Laboratorijski nalazi. Glavni laboratorijski nalaz bolesnika s KLL-om uključuje leukocitozu (ukupni broj leukocita kreće se od 50 do 500 x 109/L) uz apsolutnu limfocitozu u diferencijalnoj krvnoj slici (> 5 x 109/L). U trenu postavljanja dijagnoze kod oko 20 % bolesnika prisutna je umjerena normocitna i normokromna anemija te umjerena trombocitopenija kod oko 10%. Kada je prisutna autoimuna hemolitička anemija u laboratorijskim nalazima se očituje retikulocitoza, nekonjugirana hiperbilirubinemija te snižen haptoglobin, a potvrđuje se direktnim Coombsovim testom koji je pozitivan. Kod većine je bolesnika prisutna i hipogamaglobulinemija, dok se kod manjega broja može naći monoklonalni M-vršak u elektroforezi serumskih proteina.
Biopsija i/ili citološka punkcija koštane srži. U uzroku koštane srži, citološki gledano, dominiraju limfociti čiji je udio veći od 30 %, a kako bolest napreduje on može iznositi i 100 %. Histološki nalaz uzorka koštane srži može ispoljavati jedan od četiri oblika limfocitne infiltracije: nodularni, intersticijski, nodulointersticijski ili difuzni. U početnim fazama bolesti dominiraju prva tri tipa, no s napredovanjem bolesti razvija se difuzni oblik, a on predstavlja loš prognostički znak.
Imunofenotipizacija uzorka koštane srži ili periferne krvi omogućava otkrivanje antigenskih obilježja na površini leukemijskih stanica te tako pomaže u diferenciranju staničnog porijekla. Imunofenotipizacijom prikazuje se pozitivnost na jedan ili više B staničnih biljega (CD19, CD20, CD23) uz prisutnost biljega CD5 te restrikciju lakih lanaca imunoglobulina (kappa/lambda). Citogenetske i molekularne analize imaju više prognostički nego dijagnostički značaj u ovih bolesnika (ranije opisano).
Diferencijalna dijagnoza. Osim na limfoproliferativne bolesti, kod bolesnika s limfocitozom treba pomišljati i na virusne infekcije, kao što su pertusis, citomegalovirusna infekcija, infekciozna mononukleoza, tuberkuloza i toksolazmoza. Limfocitoza je kod tih bolesnika umjerenog i prolaznog karaktera. KLL treba razlikovati i od drugih indolentnih limfopriliferativnih bolesti, kao što su prolimfocitna leukemija, leukemija vlasastih stanica, limfom te Waldenstromova makroglobulinemija (Tablica 6.31.).
|
Tablica 6.31. Diferencijalna dijagnoza indolentnih limfoproliferativnih bolesti
|
|
Limfoproliferativna bolest
|
Limfadenopatija (%)
|
Splenomegalija (%)
|
Stanično porijeklo
|
Izražaj površinskih antigena
|
|
CD19, CD20
|
Ostali
|
|
Kronična limfocitna leukemija (KLL)
|
75
|
50
|
B
|
90 %
|
CD23, CD24
|
|
Prolimfocitna leukemija (PLL)
|
33
|
95
|
B
|
75 %
|
FMC-75
|
|
Leukemija vlasastih stanica (HCL)
|
< 10
|
80
|
B
|
> 90 %
|
CD25, CD11C
|
|
Limfom malih stanica (SCL)
|
90
|
90
|
B
|
> 90 %
|
CD10
|
|
Waldenstromova makroglobulinemija
|
33
|
33
|
B
|
> 90 %
|
CD38, PCA-1
|
Procjena proširenosti bolesti. U procjeni proširenosti bolesti koristimo se trima sustavima: Rai, Binet i TTM klasifikacija. Prema Rai klasifikaciji KLL dijelimo u pet stadija koji se numerički označavaju 0 do IV: nulti stadij (limfocitoza), prvi stadij (limfadenopatija), drugi stadij (splenomegalija, limfadenopatija ili oboje), treći stadij (anemija, organomegalija) te četvrti stadij (jedan ili više od navedenih kriterija: anemija, trombocitopenija i organomegalija). Prema Binetovoj klasifikaciji KLL se dijeli u tri stadija koja se označavaju slovima A do C: stadij A (limfocitoza uz limfadenopatiju do tri različite regije), stadij B (limfadenopatija koja zahvaća tri ili više različite regije) te stadij C (prisutnost anemije, trombocitopenije ili oboje). TTM klasifikacijom procjenjujemo ukupnu tumorsku masu. TTM klasifikacija polazi od toga da je tumorska masa raspoređena u 3 osnovna odjeljka: TM1-perifernoj krvi i koštanoj srži, TM2-limfnim čvorovima, TM3-slezeni. Veličina tumorske mase u svakom odjeljku može varirati. Veličinu tumorske mase za prvi odjeljak izračunamo iz drugog korijena apsolutnog broja limfocita u nanolitru, za drugi odjeljak izmjerimo dijametar najvećeg palpabilnog limfnog čvora u centrimetrima, a za treći odjeljak izmjerimo koliko je palpabilna slezena od lijevog rebrena luka. Navedene vrijednosti sva 3 odjeljka se zbroje i ta vrijednost čini ukupnu tumorsku masu. TTM klasifikacija razlikuje se od Raiove i Binetove klasifikacije. TTM klasifikacija mjeri veličinu tumora neovisno o insuficijenciji mijelopoeze te nam pomaže u procjeni progresije, ujedno i učinka antitumorske terapije. Očekivano preživljenje bolesnika, koji pri dijagnozi imaju veliku tumorsku masu, kraće je od onih s malom tumorskom masom. Klasifikacija TTM je dinamička klasifikacija prihvatljiva u kliničkoj praksi u svakoj fazi bolesti.
Liječenje. Većina bolesnika s KLL-om ne zahtijeva specifično liječenje, nego samo redovno praćenje i kontrole. Liječenje se započinjemo uz sljedeće indikacije: 1) manifestna mijelosupresija (teška anemija, trombocitopenija); 2) prisutnost rekurentnih infekcija; 3) masivna ili progresivna splenomegalija i/ili limfadenopatija; 4) progresija bolesti koja se manifestira udvostručenjem broja limfocita kroz šestomjesečni period; 5) izraženi sistemski simptomi (febrilno stanje, preznojavanje, gubitak na tjelesnoj težini) te 6) pojava autoimune hemolitičke anemije i/ili trombocitopenije.
Specifično liječenje podrazumijeva primjenu kemoterapeutika, a njihov izbor ponajprije ovisi o dobi i kondicijskom stanju bolesnika. Za većinu bolesnika prvu liniju liječenja čini FCR protokol (fludarabin, ciklofosfamid, rituksimab) za koji je pokazano da ima daleko bolji učinak od ranije primjenjivanih protokola. Ibrutinib, inhibitor Bruton kinaze, koristi se u prvoj liniji liječenja odraslih bolesnika s kroničnim limfocitnom leukemijom s delecijom kromosoma 17p, odnosno mutacijom p53, kod prethodno neliječenih bolesnika dobroga općeg stanja (ECOG 0-2) visokoga rizika, koji se definira prisustvom nemutiranih gena za teški lanac imunoglubulina (uIGHV), kao i za liječenje prethodno neliječenih bolesnika kod kojih nije prikladno liječenje temeljeno na punoj dozi fludarabina. Kod starijih bolesnika i bolesnika lošije kondicije prvu liniju liječenja može predstavljati i primjena bendamustina ili klorambucila s ili bez rituksimaba. U drugoj liniji liječenja na raspolaganju su ofatumumab i obinutuzumab (anti-CD20 antitijela). Osim navedenih lijekova, za liječenje refrakternih oblika, kao i relapsa bolesti, na raspolaganju su i inhibitori B-staničnih signalnih i bcl2 inhibitor (venetoklaks). Alogena transplantacija koštane srži u ovih bolesnika u obzir dolazi jedino ako se navedenim medikamentnim liječenjem ne postiže zadovoljavajući odgovor.
Nespecifično liječenje. Kod bolesnika s razvijenom autoimunom hemolitičkom anemijom i trombocitopenijom indicirana je primjena kortikosteroida. Kod bolesnika s izraženom imunosupresijom od koristi može biti primjena imunoglobulina, a preporučuje se i redovno cijepljenje protiv gripe i pneumokoka.
Prognoza. Ukupno gledano, KLL ima dobru prognozu i gotovo trećina bolesnika ima stabilnu bolest koja nikada neće zahtijevati specifično liječenje. Medijan preživljenja bolesnika u nultom stadiju bolesti iznosi više od deset godina, u prvom stadiju devet godina, drugom stadiju sedam godina te trećem i četvrtom stadiju pet godina.
LEUKEMIJA VLASASTIH STANICA
Definicija i epidemiologija. Leukemija vlasastih stanica (triholeukemija) predstavlja rijetki oblik kronične leukemije koju karakterizira pojačano stvaranje i nakupljanje zrelih limfocita (obično B-staničnog porijekla) s karakterističnim morfološkim obilježjem u obliku citoplazmatskih izdanaka nalik vlasima kose. Čini oko 2 % svih leukemija, s incidencijom od 0,3 na 100 000 osoba godišnje. Leukemija vlasastih stanica obično se javlja u petom desetljeću života, češće kod muškaraca (omjer 5:1).
Etiopatogeneza. Leukemija vlasastih stanica nastaje kao posljedica točkaste mutacije BRAF gena (V600E mutacija) smještenoga na 7. kromosomu, a što za posljedicu ima pojačanu aktivnost serin-treoinin kinaze koja aktivira unutrastanični put odgovoran za hiperproliferaciju limfocita i nastanak ove vrste leukemije. Leukemijske stanice kod ovih bolesnika na površini eksprimiraju CD19, CD20, CD11C, CD103, FMC7, CD22 antigene te receptor za IL-2. Kao rizični čimbenici za razvoj ove vrste leukemije spominju se izloženost određenim kemikalijama u poljoprivredi (pesticidi, herbicidi), petroleju i dizelu te ionizirajućem zračenju.
Klinička slika. Leukemija vlasastih stanica ubraja se u leukemije indolentnoga tijeka. Najčešći su simptomi nespecifični te uključuju umor, malaksalost i gubitak tjelesne težine. Kod više od 80 % bolesnika prisutna je splenomegalija s pridruženim simptomima (osjećaj rane sitosti i bola pod lijevim rebrenim lukom), dok su hepatomegalija i limfadenopatija rijetki. Potiskivanje stanica mijelopoeze (anemija, neutropenija, trombocitopenija) te disfunkcija stvorenih limfocita bolesnike čini izuzetno sklonima oportunističkim infekcijama, koje i predstavljaju najčešći uzrok pobola i smrtnosti. Također, ovi bolesnici imaju i povećan rizik od obolijevanja od drugih malignoma.
Dijagnoza. Sumnja na leukemiju vlasastih stanica postavlja se na temelju laboratorijskih nalaza koji uključuju leukocitozu uz limfocitozu i različito izraženu pancitopeniju te karakteristični izgled limfocita na razmazu periferne krvi. Dijagnoza se potvrđuje imunofenotipizacijom limfocita periferne krvi ili uzorka koštane srži (određivanje eksprimiranih antigena na površini stanice) uz potvrdu točkaste mutacije BRAF gena (V600E).
Liječenje je indicirano kod bolesnika s izraženim simptomima, splenomegalijom, učestalim infekcijama ili izraženom citopenijom. Prvu liniju liječenja čini primjena pentostatina ili kladribina (analozi nukleozida) uz koje se postiže kompletna remisija u više od 70 do 95 % slučajeva. Rituksimab se primjenjuje u drugoj liniji liječenja bolesnika koji ne reagiraju na primijenjenu terapiju ili imaju relaps bolesti, a kod najtežih oblika na raspolaganju stoji BRAF innhibitor vemurafenib. Uz navedeno liječenje više od 95 % bolesnika živi duže od deset godina nakon početka liječenja.
Limfom je uopćeni naziv za primarne zloćudne tumore limfatičnoga tkiva koji nastaju malignom transformacijom T-limfocita, B-limfocita ili NK-stanica. Maligna se transformacija najčešće događa u limfnim čvorovima, pa se takvi limfomi nazivaju nodalnim limfomima, iako bolest može započeti i u limfatičnom tkivu pridruženom drugim organima (koštana srž, jetra, slezena, želudac, crijeva i sl.), pa govorimo o ekstranodalnim limfomima. Oni pripadaju široj skupini bolesti koje nazivamo limfoproliferativnim bolestima, a one obuhvaćaju i leukemije i plazmastanične neoplazme jer im je svima zajedničko da potiču od limfocita, a u načelu se razlikuju prema mjestu gdje primarno nastaju (koštana srž i krv kod leukemija, odnosno limfni čvorovi i limfatično tkivo drugih organa kod limfoma) ili prema stanicama od kojih nastaju (npr. plazma stanice koje predstavljaju krajnji stadij diferencijacije B-limfocita).
Temeljna klasifikacija dijeli maligne limfome u dvije skupine: ne-Hodgkinovi i Hodgkinovi limfomi. Osnovna razlika među njima temelji se na histološkoj slici koja se razlikuje prema zastupljenosti tumorskih stanica u samome tumoru. Hodgkinovi limfomi karakterizirani su malim udjelom (obično do 5 %) tumorskih stanica unutar cijele tumorske mase koju mahom čine nespecifične, reaktivne upalne stanice, dok kod ne-Hodgkinova limfoma nalazimo obrnutu situaciju te tumorsko tkivo u najvećem dijelu čine tumorske stanice uz oskudnu prisutnost drugih stanica.
Osim toga, pojedini se tipovi Hodgkinova i ne-Hodgkinova limfoma osim prema histološkoj slici razlikuju i prema imunofenotipu (površinska ekspresija antigena), citogenetici (genetske promjene unutar tumora) te kliničkim karakteristikama (agresivnost i klinička prezentacija, odgovor na liječenje, prognoza).
Prema klasifikaciji SZO-a iz 2016., prema morfološkim, imunofenotipskim, genetičkim i kliničkim osobinama razlikuje se pet skupina limfoidnih neoplazmi: B-stanične, T- i NK-stanične, Hodgkinov limfom, neoplazme histiocita i dendritičkih stanica te poslijetransplantacijski limfoproliferacijski poremećaji. Hodgkinovi se limfomi dijele u dvije skupine:1) nodularna limfocitna predominacija te 2) klasični Hodgkinov limfom.
Kako se klasifikacija limfoma u konačnici ponajprije zasniva na porijeklu, odnosno vrsti limfocita od koje potječu, ovdje će biti opisana i normalna ontogeneza T- i B-limfocita, s obzirom na to da limfom može nastati malignom transformacijom limfocita u bilo kojoj razvojnoj fazi. Ontogenezu, odnosno razvoj limfocita prate određene promjene u pogledu ekspresije površinskih antigena (CD, od engl. cluster of differentiation) što se može dokazati različitim metodama primjene monoklonalnih antitijela (imunofenotipizacija) i to uvelike pomaže, zajedno s histološkim obilježjima i citogenetskim promjenama, u određivanju vrste limfoma.
B-limfociti imaju nekoliko stupnjeva diferencijacije: pre-B-stanice, zrele B-stanice, aktivirane/proliferirajuće B-stanice, diferencirane B-stanice i sekretorne B-stanice (plazma stanice). Antigeni CD19 i CD20 nalaze se samo na B-limfocitima i određuju njihovu linijsku pripadnost. Svaki stadij diferencijacije nosi još neke određene antigene na njihovoj površini. U procesu diferencijacije, pre-B-stanice na svojoj površini imaju CD10 (CALLA) antigen, a ubrzo se pojavljuje i CD20. Nakon toga stanice odlaze u krv i limfna tkiva gdje miruju do trenutka kada postanu aktivirane antigenom (zrele B-stanice). Zrele B-stanice na svojoj površini imaju CD19 i CD20, dok se CD10 više ne nalazi na njoj. Također mogu imati još neke antigene (CD21, CD44), kao i površinske imunoglobuline IgM i IgD. Nakon aktivacije antigenom stanice se aktiviraju i proliferiraju. Te aktivirane/proliferirajuće B-stanice gube IgD i CD21, a na površini se pojavljuju aktivacijski antigeni – CD71, CD54, CD25, CD5. U daljnjim stadijima dolazi do gubitka aktivacijskih antigena uključujući i pan-B-stanične antigene CD19 i CD20 te se pojavljuju neki novi (CD38, PCA-1) koji se nalaze na plazma stanicama.
U vrijeme embriogenoga i ranoga postnatalnog razvoja T-limfocita, stanice prekursori iz koštane srži migriraju u timus gdje mikrookoliš omogućava stvaranje funkcionalno zrelih T-limfocita. Nakon sazrijevanja u timusu T-limfociti migriraju u cirkulaciju i periferna limfna tkiva. Sazrijevanje limfocita unutar timusa praćeno je promjenama staničnih površinskih antigena. U timusu T-limfociti prolaze tri stupnja diferencijacije i u svakom iskazuju određene površinske antigene: stadij I – protimocit (CD2, CD 7, CD38, CD71), stadij II – timocit (CD1, CD2, CD4, CD7, CD8, CD38) te stadij III – timocit (CD2, CD3, CD4/8, CD5, CD6, CD7, TCR), dok u perifernoj krvi nalazimo zrelu T-pomoćničku stanicu (CD2, CD3, CD4, CD5, CD6, CD7, TCR) te zrelu T-citotoksičnu stanicu (CD2, CD3, CD4, CD5, CD6, CD7, TCR).
HODGKINOV LIMFOM (HODGKINOVA BOLEST)
Definicija i epidemiologija. Hodgkinov limfom (HL) predstavlja maligno oboljenje limfnoga tkiva B-staničnoga porijekla čije je glavno histološko obilježje nalaz karakterističnih, rijetkih Reed-Sternbergovih (RS) stanica ili njihovih mononuklearnih varijanti (Hodgkinova stanica, limfohistocitna (LH-stanica) unutar tumorskoga tkiva u najvećem postotku sastavljenoga od reaktivnih upalnih stanica i/ili fibroznoga tkiva. Hodgkinov se limfom rjeđe javlja u odnosu na ne-Hodgkinove limfome, s procijenjenom učestalošću od 3 do 5 oboljelih na 100 000 stanovnika s nešto češćom pojavnošću kod muškaraca. Pojavnost Hodgkinove bolesti prati karakteristična bimodalna raspodjela pri čemu se prvi vrh bilježi u trećem, a drugi u šestom desetljeću života, a svega se 5 % slučajeva bilježi u dobi mlađoj od 15 godina i 5 % slučajeva u dobi starijoj od 70 godina.
Etiopatogeneza. Točan uzrok i mehanizam nastanka Hodgkinova limfoma su nepoznati, a danas se njegova pojavnost najviše povezuje s infekcijom Epstein-Barrovim virusom (EBV) te s genetskom predispozicijom, iako nijedno nije u potpunosti dokazano i potvrđeno.
Infekcija Ebstein-Barrovim virusom. Na povezanost EBV-infekcije i Hodgkinova limfoma ukazuju istraživanja koja su pokazala da se kod osoba s Hodgkinovim limfomom bilježe znatno veće razine antitijela na virusni kapsidni antigen, kao i činjenica da se u više od polovice slučajeva može dokazati prisutnost virusne DNA u Reed-Sternbergovim stanicama. Ipak, ti dokazi nisu dovoljno jaki da bi se njima jasno potvrdila ta etiopatogenetska povezanost, koja i dalje ostaje nejasna.
Genetska predispozicija. Na genetsku predispoziciju kao etiološki čimbenik nastanka Hodgkinova limfoma ukazuju činjenice da se bolest s većom incidencijom javlja kod osoba čiji su rođaci u prvom koljenu također imali Hodgkinov limfom, kao i veća vjerojatnost pojave bolesti kod monozigotnoga u odnosnu na dizigotnog blizanca. Točne genetske promjene povezane s nastankom Hodgkinova limfoma nisu poznate i do kraja istražene, a kao jedna od najčešćih spominje se mutacija NPAT gena koji se nalazi na 6. kromosomu (6p21.32), čiji genski produkt djeluje kao koaktivator histonske transkripcije.
Klasifikacija. Patohistološki se razlikuju dva temeljna oblika Hodgkinova limfoma: nodularna limfocitna predominacija te klasični Hodgkinov limfom koji se dijeli u četiri podtipa koji se međusobno ne razlikuju samo prema histološkim nego i prema kliničkim i prognostičkim karakteristikama. Osnovne zloćudne stanice u tim tumorima su Reed-Sternbergove stanice te njihove varijante: mononuklearne stanice (Hodgkinove stanice) i LH-stanice, a sve imaju isto B-stanično porijeklo iz germinativnoga centra limfnoga čvora.
Nodularna limfocitna predominacija. Taj oblik Hodgkinova limfoma karakterizira nodularna ili nodularna i difuzna proliferacija malih limfocita i histiocita uz pojavu rijetkih malignih LH-stanica koje predstavljaju jednu od mononuklearnih varijanti RS-stanica. LH-stanice su velike stanice s izraženom multilobuliranom jezgrom koja liči na kokice (engl. popcorn) i s oskudnom citoplazmom, a na čijim se površinama mogu dokazati B-stanični biljezi (CD20, CD45, CD75, CD79a). Bolest se najčešće pojavljuje u cervikalnim, askilarnim i ingvinalnim limfnim čvorovima čija je struktura u potpunosti zamijenjena tumorskim tkivom. Klinički je karakterizirana sporom progresijom, čestim recidivima te dobrim odgovorom na liječenje, a u 2 do 10 % slučajeva bilježi se transformacija u difuzni B-velikostanični limfom.
Klasični Hodgkinov limfom. Taj oblik Hodgkinova limfoma karakterizira tumorska masa sastavljen od rijetkih malignih stanica (Hodgkinove stanice, RS-stanice) te reaktivni stanični infiltrat kojeg čine limfociti, eozinofili, neutrofili te druge upalne stanice. Klasične RS-stanice velike su stanice s dvije jezgre smještene jedna nasuprot drugoj u zrcalnoj simetriji, koje u svojim središtima imaju krupnu eozinofilnu jezgricu okruženu svijetlom aureolom zbog čega nastaje izgled koji se opisuje kao „sovino oko“. Mononulearna varijanta takve RS-stanice naziva se Hodgkinova stanica. Klasični Hodgkinov limfom najčešće zahvaća cervikalne i medijastinalne limfne čvorove, nešto rjeđe aksilarne i paraaortalne. Ovisno o histološkom nalazu i kliničkim karakteristikama razlikuju se četiri podtipa klasičnoga Hodgkinova limfoma: 1) nodularna skleroza, 2) mješovita celularnost, 3) limfocitna deplecija te 4) limfocitna dominacija tj. limfocitima bogat klasični Hodgkinov limfom (Tablica 6.32.).
|
Tablica 6.32. Karakteristike klasičnoga Hodgkinovog limfoma
|
|
Histološki tip i učestalost
|
Patološke karakteristike
|
Lokalizacija i prognoza
|
|
Nodularna skleroza
(40 – 80 %)
|
Lakularne RS-stanice, mješoviti upalni infiltrat, obilje vezivnog tkiva
|
Medijastinum, slezena, pluća
Vrlo dobra prognoza
|
|
Mješovita celularnost
(20 – 40 %)
|
Klasične RS-stanice, mješoviti upalni infiltrat, bez vezivnih tračaka
|
Slezena, rjeđe koštana srž i jetra
Dobra prognoza
|
|
Limfocitna deplecija
(2 – 15 %)
|
Obilje klasičnih RS-stanica, infiltrat oskudan limfocitima
|
Intraabdominalni limfni čvorovi
Loša prognoza
|
|
Limfocitna dominacija
(2 – 10 %)
|
Klasične RS-stanice, u upalnom infiltratu dominiraju mali limfociti
|
Periferni limfni čvorovi
Odlična prognoza
|
Klinička slika. Hodgkinovu bolest klinički karakterizira periferna limfadenopatija uz slabije izražene opće simptome (tzv. B-simptomi), a mogu se pojaviti i simptomi i znakovi tumorske infiltracije drugih tkiva i organa.
Periferna limfadenopatija predstavlja temeljno očitovanje Hodgkinove bolesti, a započinje povećanjem i sporim rastom jednoga ili više limfnih čvorova, najčešće cervikalne ili supraklavikularne regije. Uvećani limfni čvorovi su bezbolni, čvrste konzistencije te pomični u odnosu na podlogu. Kod manjega se broja bolesnika nakon konzumacije alkohola javi bolnost limfnih čvorova.
Opći ili B-simptomi rjeđe se pojavljuju kod bolesnika i bilježe se u oko 40 % slučajeva. Uključuju subfebrilne temperature uz noćno znojenje (Pel-Epsteinov tip temperature), gubitak na tjelesnoj težini, umor, slabost i nemoć. Često je prisutan i jak, generaliziran svrbež kože, a u nekim se slučajevima javljaju i osipi.
Ostale manifestacije. Infiltracija medijastinuma, pluća, perikarda ili pleure očituje se kašljem, bolovima u prsima i/ili dispnejom. Sindrom gornje šuplje vene uslijed medijastinalne lokalizacije tumora nekada može biti i prvi znak bolesti. Koštana se bol javlja kod infiltracije kosti. Glavobolje i smetnje vida javljaju se kod intrakranijalne diseminacije bolesti.
Dijagnoza se postavlja na osnovi anamneze, fizikalnoga pregleda, laboratorijskih nalaza te biopsije tumorske mase (limfni čvor). Kod svih bolesnika s dokazanim Hodgkinovim limfomom potrebno je obaviti dijagnostičku obradu s ciljem procjene proširenosti bolesti.
Laboratorijski nalazi su nespecifični i mogu uključivati umjerenu leukocitozu s neutrofilijom, eozinofiliju, a moguća je i limfopenija koja je povezana s lošijim ishodima i nepovoljnim histološkim tipovima. Kod jedne se trećine bolesnika javlja i anemija tipa kronične bolesti (mikrocitna, hipokromna), rjeđe autoimuna hemolitička anemija. Aktivnost bolesti prati se određivanjem vrijednosti laktat dehidrogenaze te praćenjem sedimentacije eritrocita. Kod bolesnika se mogu naći i povišene vrijednosti bakra, haptoglobina, feritina te α-2-globulina.
Biopsija i citološka punkcija limfnog čvora osnovne su dijagnostičke metode za postavljanje dijagnoze ove bolesti. Biopsijom se dobiva uzorak koji se histološki analizira, a osnovni i glavni nalaz koju upućuje na Hodgkinovu bolest je prisutnost RS-stanica i/ili njihovih mononuklearnih varijanti. Ekstirpacija cijeloga limfnog čvora predstavlja bolji izbor od same biopsije i preporuča se u svim situacijama kada je to moguće. Citološka punkcija limfnoga čvora omogućava imunofenotipizaciju kojom se ispituje ekspresija biljega na staničnoj površini, što također može imati ulogu u diferenciranju tipa Hodgkinova limfoma: nodularnu limfocitnu predominaciju karakterizira imunofenotip CD15-, CD30-, CD20+, dok klasični oblik Hodgkinova limfoma karakterizira imunofenotip CD15+, CD30+, CD20-. No, osnovu dijagoze čini patohistološka obrada ekstirpiranoga limfnog čvora. Prisutnost EBV-DNA može se dokazati u 40 do 60 % Reed-Sternbergovih stanica.
Procjena proširenosti bolesti. Procjena proširenosti bolesti. Pri procjeni proširenosti bolesti mogu se koristiti različite dijagnostičke metode uključujući pregledni radiogram srca i pluća, ultrazvuk trbuha, MSCT vrata, prsnog koša, trbuha i zdjelice te biopsija koštane srži. PET-CT danas predstavlja najjednostavniju i najsveobuhvatniju dijagnostičku metodu korištenu za procjenu proširenosti bolesti. Dostupne metode eksploracijske laparotomije ili laparoskopije s ciljem otkrivanja proširenosti bolesti danas se ne primjenjuje.
Ann Arbor klasifikacija koristi se za određivanje kliničkoga stadija bolesti, a temelji se na procjeni proširenosti tumorske mase, što ima bitnu ulogu u terapijskom pristupu bolesniku. Prema toj se klasifikaciji Hodgkinova bolest može prezentirati jednim od četiri stadija (Tablica 6.33.) uz koje se dopisuju dodatna slova ovisno o kliničkim karakteristikama: 1) za sve stadije slovo A (nema B simptoma) i B (prisutan jedan ili više B simptom) te 2) za stadije I-III slovo S (zahvaćena slezena) i E (fokalno zahvaćen ekstralimfatični organ).
|
Tablica 6.33. Ann Arbor klasifikacija
|
|
Stadij bolesti
|
Karakteristike
|
|
I stadij
|
Zahvaćena jedna regija limfnih čvorova ili jedan ekstralimfatični organ
|
|
II stadij
|
Zahvaćeno više regija limfnih čvorova ili struktura s iste strane dijafragme
|
|
III stadij
|
Zahvaćeno više regija limfnih čvorova ili struktura s obje strane dijafragme
|
|
IV stadij
|
Difuzno zahvaćanje ekstralimfatičnih organa
|
Diferencijalna dijagnoza Hodgkinove bolesti je široka i obuhvaća širi spektar bolesti, kao što su drugi oblici ne-Hodgkinova limfoma, reaktivna limfadenopatija, infekciozna mononukleoza i sl.
Liječenje. Danas osnovu liječenja Hodgkinova limfoma čini kemoterapija (Tablica 6.34.) uz zračenje zahvaćenoga polja, a sam terapijski pristup ovisi o stadiju bolesti, prisutnosti nepovoljnih prognostičkih čimbenika te dobi bolesnika.
U I. i II. stadiju bez nepovoljnih prognostičkih čimbenika primjenjuju se dva ciklusa ABVD protokola uz zračenje zahvaćenih regija dozom od 20 Gy, dok se kod bolesnika s nepovoljnim prognostičkim čimbenicima primjenjuju četiri ciklusa ABVD protokola ili dva ciklusa eBEACOPP plus dva ciklusa ABVD protokola uz zračenje zahvaćenih regija dozom od 30 Gy.
U III. i IV. stadiju kod pacijenta mlađih od 60 godina primjenjuje se šest ciklusa eBEACOPP protokola (eventualno četiri ciklusa ako je PET nakon drugoga ciklusa negativan), a kao alternativa se primjenjuje šest ciklusa ABVD protokola uz šest dodatnih ciklusa eBEACOPP, ako je PET nalaz nakon drugoga ciklusa pozitivan. Zračenje je nakon provedene kemoterapije indicirano za regije u parcijalnoj remisiji (prema nalazu PET-a) te PET negativne regije s inicijalno velikim tumorskim masama, kao i za ekstranodalne lokalizacije. Kod bolesnika u dobi od 60 do 75 godina primjenjuje se liječenje sa šest ciklusa ABVD protokola, dok se kod bolesnika starijih od 75 godina primjenjuje liječenje s dva ciklusa ABVD-a te četiri ciklusa AVD protokola.
U slučaju relapsa ili refrakternoga oblika u obzir dolazi autologna transplantacija koštane srži uz intenziviranu kemoterapiju. U slučaju povrata bolesti nakon autologne transplantacije, ili ako se radi o bolesnicima s više od dva relapsa, moguća je primjena brentuksimaba-vedotina (konjugat protutijela i lijeka koji čini monoklonsko protutijelo protiv CD30 antigena kovalentno vezano za antimikrotubularnu tvar monometil auristatin E, MMAE) i alogene transplantacije koštane srži. U slučaju neuspjeha liječenja s brentuksimabom, kao zadnja opcija ostaje liječenje nivolumabom ili pembrolizumabom (monoklonalna protutijela na receptor programirane stanične smrti).
Prognoza. Nepovoljni prognostički pokazatelji kod bolesnika s Hodgkinovom bolesti su: 1) dob iznad 45 godina, 2) hemoglobin ispod 105 g/L, 3) IV. stadij bolesti, 4) muški spol, 5) leukociti viši od 16 x 109/L, 6) limfociti niži od 0,6 x 109/L te albumini niži od 4 g/L. Stopa izlječenja kreće se oko 75 % ako su prisutna do dva nepovoljna prognostička čimbenika, odnosno do 55 % ako je prisutno tri ili više rizičnih čimbenika. Desetogodišnje preživljenje bolesnika s ranim stadijima bolesti (stadij I i II) iznosi i preko 90 %, dok je kod uznapredovalih stadija (stadij III i IV) oko 50 do 60 %.
Definicija. Ne-Hodgkinovi limfomi (NHL) predstavljaju heterogenu skupinu limfoproliferativnih bolesti koju karakterizira maligna transformacija T i B-limfocita unutar limfatičnoga tkiva, a koja se po svojim histološkim, imunofenotipskim, citogenetskim i kliničkim karakteristikama razlikuje od Hodgkinova limfoma (Tablica 6.35.). U ovom će dijelu biti opisane skupine karakteristike ne-Hodgkinova limfoma, a zatim će zasebno biti opisani najčešći oblici u kliničkoj praksi.
|
Tablica 3.35. Temeljne razlike Hodgkinova i ne-Hodgkinova limfoma
|
|
Karakteristike
|
Hodgkinov limfom
|
Ne-Hodgkinovi limfomi
|
|
Porijeklo
|
B-limfociti
|
T i B-limfociti, NK-stanice
|
|
Histološka obilježja
|
Mali udio tumorskih stanica u tumoru
|
Veliki udio tumorskih stanica u tumoru
|
|
Imunofenotipska obilježja
|
Površinski antigeni B-limfocita
|
Površinski antigeni T i B-limfocita i NK-stanica
|
|
Citogenetska obilježja
|
Rjeđa specifična citogenetska obilježja
|
Češća specifična citogenetska obilježja
|
|
Lokalizacija
|
Aksijalni limfni čvorovi
|
Aksijalni i ekstraaksijalni limfni čvorovi
|
|
Širenje bolesti
|
Kontinuirano
|
Diskontinuirano
|
|
Ekstranodalna pojavnost
|
Rijetka
|
Česta
|
Epidemiologija. Dok je učestalost Hodgkinova limfoma prilično stabilna, učestalost ne-Hodgkinovih limfoma u stalnome je porastu i njegova godišnja incidencija danas iznosi oko 15 do 20 oboljelih na 100 000 stanovnika. Kod većine oblika ne-Hodgkinovih limfoma učestalost raste s dobi i najviša je u petom desetljeću života. Bolest se nešto češće javlja kod muškaraca u odnosu na žene. Zanimljiva je i zemljopisna raspodjela ove bolesti jer su ne-Hodgkinovi limfomi B-staničnoga porijekla češći u zemljopisnim krajevima s umjerenom klimom (oko 85 % slučajeva), dok u tropskim krajevima prevladavaju tumori T-staničnoga porijekla.
Etiopatogeneza ne-Hodgkinovih limfoma nije u potpunosti poznata, ali ulogu u njihovom nastanku i razvoju mogu imati genetski, okolišni i imunološki čimbenici.
Genetski čimbenici. Danas se zna da je veliki dio pojedinih oblika ne-Hodgkinova limfoma povezan sa specifičnim genetskim promjenama koje se povezuju s malignom transformacijom limfatičnih stanica. Te se promjene odnose na specifične kromosomske translokacije i posljedičnu abnormalnu ekspresiju onkogena, tj. gena koji potiču malignu transformaciju i tumorski rast. Neke od najznačajnijih citogenetskih promjena u ovoj skupini bolesti prikazane su u Tablici 6.36.
|
Tablica 6.36. Najčešće citogenetske promjene kod ne-Hodgkinovih limfoma
|
|
Vrsta non-Hodgkinova limfoma
|
Citogenetski poremećaj
|
Zahvaćeni geni
|
|
Difuzni B-velikostanični limfom
|
t(3q27)
|
BCL6
|
|
t(14;18)(q32;q21)
|
IgH, BCL2
|
|
t(8;14)(q24;q32)
|
MYC, IgH
|
|
Burkittov limfom
|
t(8;14)(q24;q32)
|
MYC, IgH
|
|
t(8;22)(q24;q11)
|
MYC, IgL
|
|
t(2;8)(p12;q24)
|
IgK, MYC
|
|
Folikularni limfom
|
t(14;18)(q32;q21)
|
IgH, BCL2
|
|
Limfom plaštene zone
|
t(11;14)(q13;q32)
|
BCL1, IgH
|
|
Anaplastični velikostanični limfom
|
t(2;5)(p23;q35)
|
ALK, NPM
|
Okolišni čimbenici. Epidemiološke studije dokazale su povećanu učestalost ne-Hodgkinovih limfoma kod osoba koje su bile izložene određenim okolišnim čimbenicima od kojih su najznačajniji zračenje i virusne infekcije, osobito Epstein-Barrovim virusom i humanim T-leukemijskim virusom. Primjena određenih lijekova, kao što su fenitoin i alkilirajući lijekovi, također može biti povezana s povećanom pojavnosti ove skupine bolesti.
Imunološki čimbenici. Činjenica da je pojavnost ne-Hodgkinova limfoma viša kod bolesnika s autoimunim bolestima i sindromima prirođene ili stečene imunodeficijencije, kao i kod bolesnika koji primaju imunosupresivnu terapiju kroz dulji vremenski period, sugerira da imunološki sustav ima određenu ulogu u nastanku ovih bolesti.
Klasifikacija. Histološka podjela tumora limfocitne loze temelji se na porijeklu tumorskih stanica: 1) neoplazme prekursorskih T i B-stanica, 2) neoplazme zrelih B-stanica te 3) neoplazme zrelih T i NK-stanica. Prema stupnju malignosti ne-Hodgkinove limfome dijelimo u tri skupine: limfomi niskoga stupnja malignosti, limfomi srednjega stupnja malignosti i limfomi visokoga stupnja malignosti, dok se prema kliničkim osobinama dijele na indolentne, agresivne i vrlo agresivne limfome. Određivanje tipa ne-Hodgkinova limfoma vrši se na temelju histoloških, imunofenotipskih te citogenetskih obilježja tumora. Službenu klasifikaciju tumora limfocitne loze stvorila je Svjetska zdravstvena organizacija, a ona se odnosi na sve oblike limfoproliferativnih bolesti, pa uključuje i leukemije (limfoproliferativne bolesti koje primarno nastaju u koštanoj srži i krvi) te plazmastanične neoplazme. Navedena klasifikacija prikazana je u Tablici 6.37. U ovome dijelu teksta bit će opisivani klinički entiteti limfoma, dok su limfocitne leukemije i plazmastanične neoplazme opisane zasebno (akutne leukemije, kronične leukemije, plazmastanične neoplazme).
|
Tablica 6.37. Najčešći oblici tumora limfocitne loze prema klasifikaciji SZO-a
|
|
Neoplazme prekursorskih stanica T i B
|
Neoplazme zrelih B-stanica
|
Neoplazme zrelih T i NK-stanica
|
|
Prekusorska T-limfoblastična leukemija/ limfoblastični limfom
Prekusorska B-limfoblastična leukemija/ limfoblastični limfom
|
Kronična limfocitna leukemija/limfom malih stanica
Limfom marginalne zone
Leukemija vlasastih stanica
Folikularni limfom
Difuzni B-velikostanični limfom
Limfom plaštenih stanica
Burkittov limfom
Plazmastanične neoplazme
|
Prolimfocitna leukemija T-stanica
T-stanična leukemija velikih granuliranih limfocita
T-stanična leukemija/limfom odraslih
Agresivna NK-stanična leukemija
Anaplastični velikostanični limfom
Mycosis fungoides
Sezaryjev sindrom
|
Klinička slika. Ne-Hodgkinov limfom može se prezentirati simptomima i znakovima limfadenopatije, općim simptomima ili simptomima ekstranodalne lokalizacije bolesti. Klinički tijek bolesti u prvom redu ovisi o prirodi tumora, o tome je li indolentan, agresivan ili vrlo agresivan (Tablica 6.38.).
|
Tablica 6.38. Podjela ne-Hodgkinovih limfoma prema kliničkom karakteru
|
|
Indolentni ne-Hodgkinovi limfomi
|
Agresivni ne-Hodgkinovi limfomi
|
Vrlo agresivni ne-Hodgkinovi limfomi
|
|
Kronična limfocitna leukemija/limfom malih stanica
Limfoplazmocitoidni limfom
Limfom marginalne zone
Leukemija vlasastih stanica
Folikularni limfom
T-stanična leukemija velikih granuliranih limfocita
Mycosis fungoides
|
Limfom plaštenih stanica
Difuzni B-velikostanični limfom
Prolimfocitna leukemija T-stanica
Sezaryjev sindrom
Anaplastični velikostanični limfom
|
Prekusorska T-limfoblastična leukemija/ limfoblastični limfom
Prekusorska B-limfoblastična leukemija/ limfoblastični limfom
Burkittov limfom
Agresivna NK-stanična leukemija
|
Limfadenopatija. Najčešća klinička manifestacija ne-Hodgkinova limfoma je periferna limfadenopatija (veličina limfnih čvorova > 1 cm, odnosno > 1,5 cm u preponama), obično u području vrata ili prepona, pri čemu su zahvaćeni limfni čvorovi uvećani, bezbolni i čvršće konzistencije i često međusobno konfluiraju. Kod bolesnika nije rijetka niti medijastinalna ili retoroperitonealna limfadenopatija pri čemu se bolesnici mogu žaliti na specifične tegobe kao što su bolovi u prsima, otežano disanje, otežano gutanje, promuklost, bol u leđima i slabinama i sl. Najteži oblici medijastinalne limfadenopatije prezentiraju se sindromom gornje šuplje vene.
Opći simptomi. Simptomi i znakovi bolesti mogu biti opći ili uzrokovani samom tumorskom masom. Opći su simptomi, tzv. B simptomi, paraneoplastički simptomi uzrokovani otpuštanjem citokina iz tumorskih stanica ili stanica imunosnoga sustava koje reagiraju na tumor. Oni uključuju neobjašnjiv gubitak više od 10 % tjelesne mase u posljednjih šest mjeseci, neobjašnjivu vrućicu > 38 C i profuzno noćno znojenje. Opći B simptomi pojavljuju se u 30 % slučajeva. Simptomi uzrokovani samom tumorskom masom ovise o veličini i lokalizaciji.
Ekstranodalne manifestacije. Kako je navedeno, ove vrste limfoma imaju sklonost zahvaćanja i limfatičnoga tkiva pridruženoga drugim organima. Najčešća ekstranodalna lokalizacija je želudac/probavni sustav, ali se bolest može pojaviti u bilo kojem organu, uključujući testis, kost, pluća, štitnjaču, žlijezde slinovnice, krajnike, kožu, jetru, dojke, nadbubrežne žlijezde, bubreg, nosnu šupljinu, paranazalne sinuse, vrat maternice, rodnicu i središnji živčani sustav. Zbog toga se oni mogu očitovati i neurološkim simptomima (limfomi mozga), dispnoičnim tegobama i bolovima u prsima (limfom pluća), probavnim smetnjama i manifestacijama (limfom želuca i crijeva), pancitopenijom i njezinim posljedicama (diseminacija u koštanu srž), testikularnom masom i sl.
Dijagnoza. Dijagnostički postupak se kod takvih bolesnika sastoji od dva dijela. U prvome je dijelu bitno postaviti dijagnozu i odrediti vrstu ne-Hodgkinova limfoma, a potom je bitno procijeniti proširenost bolest s obzirom na to da ona ima utjecaj na izbor terapijskoga pristupa.
Postavljanje dijagnoze. S obzirom na nespecifičnosti kliničkoga pregleda i laboratorijskih nalaza, ključan korak u postavljanju dijagnoze je citološka punkcija ili biopsija tumorske mase (limfnoga čvora ili pridruženoga limfatičnog tkiva). Osim citološkoga ili histološkog pregleda uzorka obvezno je provesti i imunofenotipizaciju, kao i citogenetsku analizu, kako bi se olakšala diferencijacija tumora. Biopsija je (preferira se operacijska ekstripacija limfnoga čvora u cijelosti u odnosu na iglenu biopsije) zlatni standard u pogledu postavljanja dijagnoze. Citološka aspiracija može se koristiti kao test probiranja ili eventualno za potvrdu relapsa bolesti. Citologiju ne treba koristiti za postavljanje inicijalne dijagnoze, osim u slučajevima leukemijskoga oblika bolesti - kada je treba nadopuniti imunofenotipizacijom leukemijskih stanica na protočnom citometru te obično i citogenetskom i molekulskom analizom. Ako se radi o limfocitnim leukemijama, dijagnoza se postavlja na temelju analize uzorka koštane srži.
Procjena proširenosti. Zlatni standard za procjenu proširenosti limfoma (kod agresivnih limfoma) predstavlja kombinacija kompjuterizirane tomografije i pozitronske emisijske tomografije, tzv. PET-CT. CT omogućava dobar morfološki prikaz uvećanih limfnih čvorova (> 1 cm), dok se primjenom PET metode prati pojačana metabolička aktivnost koju posjeduje tumorsko tkivo. Kombinacija tih tehnika dolazi do izražaja kod limfoma koje nalazimo i u limfnim čvorovima manjim od 1 cm, kao i u praćenju liječenja kada bolest ne mora biti prisutna čak ni u limfnim čvorovima većim od 1 cm. Kod indolentnih limfoma koristi se kompjuterizirana tomografija. Za utvrđivanje limfoma središnjega živčanog sustava najčešće se koristi MR mozga uz MR spektroskopiju. Diseminacija bolesti u koštanu srž procjenjuje se na temelju biopsije kosti. Klasični rendgenogram srca i pluća, kao i ultrazvuk abdomena, koriste se, ali nisu dovoljno osjetljivi pa im je svrha u većoj mjeri isključivo orijentacijska. Procjena proširenosti bolesti indicirana je samo kod bolesnika koje se planira aktivno liječiti, dok procjena kod bolesnika koje se liječi palijativno nije potrebna (kao ni kod bolesnika s dijagnosticiranom leukemijom). Stupanj proširenosti bolesti definira se Ann Arbor klasifikacijom, kao i kod Hodgkinova limfoma (opisano u poglavlju o Hodgkinovu limfomu).
Liječenje bolesnika s ne-Hodgkinovom bolesti sastoji se od kemoterapije, radioterapije, imunoterapije te radioimunoterapije. Ovdje će biti prikazani uopćeni principi liječenja, a detaljnije će o liječenju pojedinih tipova ne-Hodgkinova limfoma biti govora u nastavku teksta.
Kemoterapija je osnovni i najčešći oblik liječenja ne-Hodgkinovih limfoma s obzirom na to da se radi o tumorima koji pokazuju dobru osjetljivost na kemoterapeutike, tj. njihove kombinacije. Izbor kemoterapijskoga protokola ponajprije ovisi o agresivnosti tumora, a navedeni su najčešći primjeri.
Najčešći kemoterapijski protokoli za liječenje indolentnih ne-Hodgkinovih limfoma su: 1) klorambucil (kontinuirana primjena visokih, standardnih ili malih doza obično u kombinaciji s glukokortikoidima); 2) Paul-Ehrlich protokol (opetovana primjena klorambucila i prednizona svaka tri do četiri tjedna po sedam dana); 3) CVP protokol (ciklofosfamid, vinkristin, prednizon svaka tri tjedna); 4) primjena fludarabina s ili bez prednizona svaka tri do četiri tjedna; 5) FC protokol (fludarabin, ciklofosfamid svaka tri do četiri tjedna); 6) FND protokol (fludarabin, mitoksantron, deksametazon svaka četiri tjedna) te 7) primjena bendamustina svaka tri do četiri tjedna.
Najčešći kemoterapijski protokoli za liječenje agresivnih ne-Hodgkinovnih limfoma u prvoj liniji su: 1) CHOP protokol (ciklofosfamid, doksorubicin, vinkristin, prednizon svaka dva do tri tjedna uz faktor rasta granulocita); 2) CHEOP protokol (CHOP + etopozid svaka dva do tri tjedna uz faktor rasta granulocita); 3) CEOP protokol (ciklofosfamid, etopozid, vinkristin, prednizon svaka tri tjedna); 4) CNOP protokol (ciklofosfamid, mitoksantron, vinkristin, prednizon svaka tri tjedna) te 5) (DA)-EPOCH protokol (etopozid, doksorubicin, vinkristin, ciklofosfamid, prednizon svaka tri tjedna uz faktor rasta granulocita od šestoga dana do oporavka granulocita).
Najčešći kemoterapijski protokoli za liječenje agresivnih ne-Hodgkinovnih limfoma u drugoj liniji jesu: 1) DHAP protokol (deksametazon, citarabin, cisplatina svaka tri do četiri tjedna); 2) ESHAP protokol (etopozid, cisplatina, citarabin, metilprednizolon svaka tri do četiri tjedna); 3) IMVP protokol (ifosfamid, uromiteksan, metotreksat, etopozid svaka tri do četiri tjedna) te 4) ICE protokol (ifosfamid, uromiteksan, karboplatina, etopozid svaka tri do četiri tjedna uz faktor rasta granulocita).
Kemoterapija primarnoga limfoma središnjega živčanog sustava sastoji se od visoke doze metotreksata s ili bez primjene citarabina uz primjenu folne kiseline uz eventualno primjenu tiotepe.
Radioterapija se koristi u svrhu lokalne kontrole bolesti. Najčešće indikacije za njezinu primjenu su lokalizirana bolest, masivna bolest (limfni čvorovi promjera većeg od 7 cm) te zaostaci tumorske mase nakon provedene kemoterapije. Zračenje se zasniva na kompjuterskom trodimenzionalnom planiranju zbog što preciznijega određivanja mjesta zračenja i poštede okolnoga tkiva. Doze zračenje iznose od 20 Gy za indolentne do 40 Gy za agresivne ne-Hodgkinove tumore.
Imunoterapija podrazumijeva primjenu monoklonskih protutijela koja su usmjerena na specifične antigene smještene na površini tumorskih stanica za koje se vežu te ostvaruju svoj citotoksičan učinak. Od 70-ih godina prošloga stoljeća, kada su antraciklini uvedeni u terapiju, pa sve do početka ovoga stoljeća nije bilo značajnijega napretka u liječenju NHL-a. Najveći napredak u liječenju B-staničnih NHL-a od uvođenja kombinirane kemoterapije ostvaren je dodavanjem monoklonskoga protutijela protiv antigena CD20 (rituksimab - R). Ovi se lijekovi mogu primjenjivati samostalno kod nekih oblika indolentnih ne-Hodgkinovih limfoma ili zajedno s kemoterapijom (R-CVP, R-CHOP, R-EPOCH). Temeljni lijek iz ove skupine je rituksimab, kimerno monoklonalno antitijelo tipa I usmjereno na CD20 antigen koji se nalazi samo na tumorima B-staničnoga porijekla. Noviji lijek iz te skupine je obinutuzumab koji pretstavlja humanizirano anti-CD20 monoklonsko protutijelo tipa II iz podskupine IgG1, bolje ga se podnosi od kimernoga (manje je imunogen). Proizveden je s ciljem poboljšanja terapije za pacijente s kroničnom limfocitnom leukemijom i B-staničnim limfomom. Osim monoklonih protutijela, postoje i konjugati protutijela i lijeka. Jedan od njih je i brentuksimab vedotin (konjugat protutijela i lijeka koji čini monoklonsko protutijelo protiv CD30 stanica kovalentno vezano za antimikrotubularnu tvar monometil auristatin E - MMAE) koji je usmjeren na CD30 antigen te se koristi u liječenju CD30+ ne-Hodgkinovih limfoma (u drugoj liniji liječenja), kao i Hodgkinova limfoma (treća linija liječenja). Polatuzumab vedotin je drugi konjugat protutijela i lijeka, a sastoji se od antimitotskoga lijeka monometilauristatina E (MMAE) kovalentnom vezom konjugiranoga s monoklonskim protutijelom koje ciljano djeluje na CD79b. Primjenjuje se u kombinaciji s bendamustinom i rituksimabom za liječenje odraslih bolesnika s relapsnim/refraktornim difuznim B-velikostaničnim limfomom koji nisu kandidati za presađivanje hematopoetskih matičnih stanica. Noviji način liječenja predstavljaju i CAR-T stanice koje predstavljaju oblik imunostanične terapije koja sadrži autologne T-stanice genetički modificirane ex vivo lentivirusnim vektorom koji kodira anti-CD19 kimerični antigenski receptor (engl. chimeric antigen receptor, CAR). Indiciran je u liječenju pedijatrijskih bolesnika te mladih odraslih bolesnika u dobi do 25 godina s akutnom limfoblastičnom leukemijom B-stanica (ALL) koja je refraktorna, u recidivu nakon transplantacije, ili u drugom ili kasnijem recidivu, kao i za liječenje odraslih bolesnika s recidivirajućim ili refraktornim difuznim B-velikostaničnim limfomom (DLBCL) nakon dvije ili više linija sistemske terapije.
Praćenje bolesnika. Pri praćenju bolesnika nakon započetoga liječenja možemo očekivati nekoliko ishoda: kompletnu remisiju, nepotvrđenu kompletnu remisiju, parcijalnu remisiju, stabilnu bolest te progresivnu bolest. U svrhu praćenja bolesti preporuča se PET-CT tehnika pri čemu se procjenjuje metabolička aktivnost bolesti prije i nakon liječenja. Ako se za praćenje u svrhu procjene remisije koristi samo CT, prati se veličina limfnih čvorova (svi čvorovi koji su prije terapije bili veći od 1,5 cm trebaju se smanjiti na 1,5 cm ili manje, a svi čvorovi koji su prije terapije bili između 1 i 1,5 cm trebaju biti manji od 1 cm).
NEOPLAZME PREKURSORSKIH STANICA T I B
Prekursorski limfoblastični T i B-limfom su neoplazme nastale malignom transformacijom nezrelih oblika limfatičnih stanica s ishodištem u limfnim čvorovima, za razliku od prekursorske limfoblastične T i B-leukemije čije je ishodište u koštanoj srži. Pojavnost tih oblika limfoma veća je u mlađoj životnoj dobi. Prekursorski B-limfoblastični limfom najčešće se klinički prezentira kao solidna masa koja zahvaća kožu i kosti, dok se prekursorski T-limfoblastični limfom obično prezentira kao medijastinalna masa. Kod tih je oblika limfoma česta pojava i infiltracija središnjega živčanog sustava, što ujedno predstavlja i loš prognostički znak. Liječenje je istovjetno liječenju akutnih limfoblastičkih leukemija uz provođenje profilakse diseminacije bolesti u središnji živčani sustav (intratekalna kemoterapija metotreksta, citarabin, ozračivanje neurokranija).
NEOPLAZME ZRELIH B-STANICA
Limfom malih stanica predstavlja limfoproliferativnu bolest koja nastaje malignom transformacijom zrelih B-limfocita unutar limfnoga čvora, čije su tumorske stanice identične onima kakve nalazimo pri kroničnoj limfatičnoj leukemiji (KLL), no broj atipičnih limfocita u perifrenoj krvi manji je od 5 x 109. Radi se o indolentnom limfomu koji se najčešće prezentira bezbolnom limfadenopatijom i/ili splenomegalijom. Prisutnost B simptoma je rijetka. U laboratorijskim nalazima česta je hiper- ili hipogamaglobulinemija. Loša prognoza povezuje se s prisutnošću B simptoma, izražajem površinskih antigena CD38 i ZAP70 te genetskim abnormalnostima del(17p) i del(11q). Kod oko 10 % bolesnika prati se transformacija u difuzni B-velikostanični limfom što se naziva Richterovim sindromom i ima lošu prognozu. Liječenje je indicirano kod svih bolesnika koji imaju prisutne B simptome, pancitopeniju ili brzoprogresivnu perifernu limfadenopatiju i/ili splenomegaliju. Liječenje mlađih bolesnika najčešće obuhvaća RFC protokol (ritukimab, fludarabin, ciklofosfamid), a kod starijih primjenu bendamustina i rituksimaba. Ostale terapijske mogućnosti uključuju primjenu imunomodulatora lenalidomida ili inhibitora B-staničnih receptora (ibrutinib, idelalisib) ili venetoklaksa kao selektivnoga inhibitora antiapoptotskoga proteina B-staničnoga limfoma 2 (bcl 2). U obzir dolazi i transplantacija krvotvornih matičnih stanica. Medijan preživljenja većine bolesnika iznosi preko 10 godina, osobito bolesnika bez loših prognostičkih znakova - njih se inicijalno ne mora liječiti, dovoljno ih je redovito pratiti.
Folikularni limfom najčešći je indolentni limfom u kliničkoj praksi. Tumorsko tkivo čine brojne nakupine malih limfocita i transformiranih stanica koje nazivamo centrocitima i centroblastima zbog čega se formira folikularna slika nalik na normalnu strukturu limfnoga čvora. Ovisno o broju velikih stanica (centroblasta), folikularni se limfom dijeli u tri stupnja (prvi, drugi i treći stupanj (a, b). Većina bolesnika je asimptomatska, a bolest se prezentira bezbolnom limfadenopatijom. Medijastinalni limfni čvorovi i slezena rjeđe su zahvaćeni, a rjeđe se susreću i B simptomi. Liječenje se indicira kod bolesnika s proširenom bolesti, prisutnošću B simptoma, kao i kod svih bolesnika s folikularnim limfomom trećega stupnja. Prvu liniju liječenja obično čini primjena kemoterapije i rituksimaba/kemoterapija ili obinutuzumab (R/O-CHOP, R/O-CVP, R/O-B) uz terapiju održavanja monoklonskim protutijelom kroz dvije godine, dok se kod relapsa i refrekternih oblika može primjenjivati i transplantacija autolognih krvotvornih matičnih stanica. Petogodišnje preživljenje prisutno je kod više od 70 % oboljelih, a prosječno preživljenje (medijan) iznosi devet godina od postavljanja dijagnoze.
Difuzni B-velikostanični limfom najčešći je oblik NHL-a. Karakteriziran je izraženom morfološkom i genetskom heterogenošću, a tri najčešća oblika su nespecificirani difuzni B-velikostanični limfom, B-velikostanični limfom bogat T-stanicama ili histiocitima te medijastinalni B-velikostanični limfom. Ova vrsta limfoma može nastati de novo - pa govorimo o primarnom obliku, ili nastaje transformacijom indolentnih limfoma, kao što je limfom malih stanica ili folikularni limfom, pa govorimo o sekundarnom obliku. Klinički se bolest može očitovati perifernom i/ili visceralnom limfadenopatijom uz izraženije B simptome u odnosu na druge oblike limfoma. Osim nodalne lokalizacije, taj oblik limfoma može imati i ekstranodalne lokalizacije, najčešće u probavnom sustavu, ali i u koži, središnjem živčanom sustavu, kostima, testisima, žlijezdama slinovnicama, plućima, bubrezima i jetri. Prvu liniju liječenja čini CHOP protokol, ili njemu slični protokoli (DA-EPOCH, CHOEP) uz dodatak rituksimaba, a kod bolesnika s čimbenicima rizika za širenje bolesti u središnji živčani sustav (zahvaćenost dojke, paranazalnih sinusa, retroperitoneuma, koštane srži i Waldeyerova prstena) preporuča se i provođenje kemoterapijske profilakse (6 - 8 intratekalnih aplikacija metotreksata ili dva ciklusa sistemske primjene visokih doza metotreksta kao dodatak R-CHOP protokolu). Druga linija liječenja (kod bolesnika koji ne postižu remisiju ili bolesnika s relapsom bolesti) provodi se neki od prethodno navedenih protokola za liječenje agresivnih NHL-a (DHAP, HDIM, ESHAP, ICE) uz primjenu rituksimaba, uz uvjet da je od njegove zadnje primjene prošlo više od šest mjeseci, te autologna transplantacija koštane srži. Polatuzumab vedotin primjenjuje se u kombinaciji s bendamustinom i rituksimabom za liječenje odraslih bolesnika s relapsnim/refraktornim difuznim B-velikostaničnim limfomom koji nisu kandidati za presađivanje hematopoetskih matičnih stanica. CAR-T imunostanična terapija koja sadrži autologne T-stanice genetički modificirane ex vivo lentivirusnim vektorom koji kodira anti-CD19 kimerični antigenski receptor (engl. chimeric antigen receptor, CAR) indicirana je za liječenje odraslih bolesnika s recidivirajućim ili refraktornim difuznim B-velikostaničnim limfomom (DLBCL) nakon dvije ili više linija sistemske terapije. Ukupno petogodišnje preživljenje bolesnika iznosi oko 60 %.
Limfom plaštenih stanica (engl., mantle cell lymphoma) građen je od srednje velikih stanica nalik centrocitima koje na svojoj površini eksprimiraju antigenske strukture CD5, FMC7, D43, BCL2 te ciklin-D1. Kod većine bolesnika citogenetskim ispitivanjima dokazuje se t(11;14)(q13:q32). Uglavnom se radi o limfomu koji se javlja u starijoj životnoj dobi i u trenutku postavljanja dijagnoze obično već pokazuje znakove diseminacije. Iako po svojim osobinama pripada skupini agresivnih limfoma, kod nekih se bolesnika može ponašati i indolentno. Liječenje uključuje primjenu alterirajućega protokola R-CHOP/ R-DHAP nakon čega slijedi autologna transplantacija koštane srži. a potom terapija održavanja ritukismabom svaka dva mjeseca kroz tri godine. U prvoj liniji liječenja kod starijih ljudi može se koristiti rituksimab uz bendamustin. U drugoj liniji dostupni su ibrutinib, lenalidomid, bortezomib. Svi navedeni lijekovi mogu se koristiti samostalno ili u kombinaciji. Petogodišnje preživljenje ovih bolesnika kreće se oko 40 %.
Burkittov limfom je izrazito agresivna neoplazma koja se obično pojavljuje ekstranodalno ili pokazuje sliku akutne leukemije građene od monoformnih, srednje velikih limfocita B. Javlja se u tri oblika: 1) endemski oblik u Africi (češći kod djece, povezan s infekcijom Epstein-Barrovim virusom), 2) sporadični oblik te 3) Burkittov limfom imunodeficijentnih bolesnika (HIV-infekcija). Česta je kromosomska aberacija t(8;14)(q24;q.32). Burkittov limfom klinički karakterizira izrazito brz rast te se u vrijeme postavljanja dijagnoze već radi o diseminiranoj bolesti koja se prezentira velikom intraabdominalnom tumorskom masom, a česta je i afekcija moždanih ovojnica. Liječenje treba započeti što ranije s obzirom na brzi rast tumora, u današnje se doba preporučuje primjena DA-R-EPOCH protokola. Alternativa je intenzivirana kemoterapija utemeljena na visokim dozama metotreksata i rituksimaba u više faza uz obveznu profilaksu širenja tumora u središnji živčani sustav (intratekalna kemoterapija ili sistemska primjena visokih doza metotreksata). U slučaju pojave relapsa jedinu terapijsku opciju predstavlja alogena transplantacija koštane srži. Petogodišnje preživljenje ovih bolesnika iznosi oko 80 %.
NEOPLAZME ZRELIH T I NK-STANICA
T-stanična leukemija/limfom odraslih pripada skupini perifernih T-staničnih tumora građenih od zrelih limfocita T, a etiološki se povezuju s infekcijom humanim T-leukemijskim virusom tipa 1 (HTLV-1). Bolest se klinički može manifestirati kao akutna leukemija, agresivni ili indolentni limfom. U agresivnoj se formi javljaju generalizirana limfadenopatija, splenomegalija, kožne infiltracije te hiperkalcijemija. Bolesnici s indolentnom formom ne zahtijevaju liječenje, dok se u slučaju agresivnih formi primjenjuje liječenje kemoterapijom (najčešće CHOP protokol) s ili bez alogene transplantacije koštane srži.
Anaplastični velikostanični limfom predstavlja agresivni limfom porijekla zrelih T-stanica koji se obično susreće kod mlađih muškaraca, a karakteriziran je kromosomskom translokacijom t(2;5) koja dovodi do prekomjerne ekspresije ALK proteina. ALK negativni anaplastični velikostanični limfomi su rijetki i imaju lošiju prognozu. Bolest karakterizira generalizirana limfadenopatija, ekstranodalna pojavnost (koža, kosti, probavni sustav) te prisutnost B simptoma. Taj se oblik limfoma danas povezuje s implantatima dojke. Osnovu liječenja čini CHOP protokol s ili bez dodatka etopozida, a autologna transplantacija koštane srži dolazi u obzir kod bolesnika u relapsu. Osim toga se u liječenju mogu primijeniti i brentuksimab vedotin (anti-CD30 antitijelo) te cirzotinib (ALK inhibitor).
Mycosis fungoides je indolentni limfom zrelih T-stanica koji primarno zahvaća kožu, dok leukemični oblik fungoidne mikoze nazivamo Sezaryjev sindrom. Mycosis fungoides javlja se kod osoba srednje i starije životne dobi, a klinički prolazi kroz nekoliko faza: eritematozna faza, faza plaka te faza tumora. Kasnije se u tijeku bolesti može javiti i periferna i visceralna limfadenopatija. Lokalizirani oblik bolesti liječi se zračenjem cijeloga tijela elektronima (TSEB, engl. total skin electron beam) uz lokalnu ili sistemsku primjenu citostatika. Sistemska kemoterapija primjenjuje se pri proširenoj bolesti (gemcitabin, fludarabin, retinoična kiselina, alfa interferon). Brentuksimab vedotin je indiciran za liječenje odraslih bolesnika s CD30+ kožnim limfomom T-stanica (CTCL) nakon najmanje jedne prethodne sistemske terapije.
Uvod. Plazmastanične neoplazme predstavljaju heterogenu skupinu bolesti karakteriziranih klonalnom proliferacijom plazma stanica s posljedičnim pojačanim izlučivanjem imunoglobulina ili njihovih dijelova (laki ili teški lanci), a taj stvoreni imunoglobulinski produkt nazivamo monoklonalni vršak ili M-komponenta koju se jasno uočava na nalazu elektroforeze serumskih proteina. Najznačajnije plazmastanične neoplazme su monoklonalna gamapatija neodređenoga značenja, multipli mijelom, Waldenstromova makroglobulinemija, bolest teških lanaca te krioglobulinemija.
MONOKLONALNA GAMAPATIJA NEODREĐENOGA ZNAČENJA
Definicija. Monoklonalna gamapatija neodređenoga značenja (MGUS, engl. monoclonal gammopathy of undetermined significance), ranije zvana benigna monoklonalna gamapatija, označava premaligni klonalni poremećaj plazma stanica obilježen prisutnošću M-komponente u nalazu elektroforeze serumskih proteina kod bolesnika koji nemaju kriterije za multipli mijelom, makroglobulinemiju, amiloidozu ili sličan poremećaj. Glavni klinički značaj ovoga poremećaja leži u mogućnosti njegove preobrazbe u multipli mijelom, makroglobulinemiju ili drugu B-staničnu neoplazmu, što se događa kod oko 25 % bolesnika (oko 1 % godišnje).
Epidemiologija. Smatra se da više od 50 % bolesnika s detektiranom prisutnošću M-komponente ima taj poremećaj. Ukupna prevalencija kod odraslih osoba iznosi oko 3 % i ovisi o dobi, tako da ona između petoga i sedmog desetljeća života iznosi oko 1 %, dok je kod starijih oko 5 %. Učestalost toga poremećaja dva je puta veća kod muškaraca nego kod žena.
Etiopatogeneza. Monoklonalna gamapatija neodređenoga značenja nastaje kao posljedica monoklonalne ekspanzije plazma stanica koje ne pokazuju obrazac malignoga ponašanja. Točan uzrok i mehanizam nastanka nisu poznati, a kao rizični čimbenici navode se starija životna dob, muški spol, obiteljska pojavnost, imunosupresija te duža izloženost pesticidima. Kod oko polovice bolesnika mogu se naći strukturne ili brojčane promjene kromosoma 14 u području lokusa za teški lanac imunoglobulina (IGH gen).
Klinička slika i dijagnoza. Bolest je obično asimptomatske naravi, pa se i dijagnosticira na temelju slučajno otkrivenih patoloških laboratorijskih nalaza (M-komponenta u serumu ili urinu). M-komponentu kod takvih bolesnika najčešće čine imunoglobulin G, imunoglobulin M ili imunoglobulin A (80 %), dok kod oko 20 % bolesnika M-komponentnu čine samo laki lanci imunoglobulina. Glavni dijagnostički kriteriji za postavljanje dijagnoze monoklonalne gamapatije neodređenoga značenja prema Svjetskoj zdravstvenoj organizaciji su: 1) M-komponenta u serumu < 30 g/L; 2) < 10 % plazma stanica u koštanoj srži; 3) odsutnost anemije, hiperkalcijemije, osteolitičkih lezija i bubrežne disfukcije te 4) isključene druge limfoproliferativne bolesti B-staničnoga porijekla.
Liječenje i prognoza. Specifično liječenje bolesnika s monoklonalnom gamapatijom neodređenoga značenja nije potrebno. Svim se bolesnicima preporuča periodično (svakih šest do dvanaest mjeseci) mjerenje koncentracije M-komponente u serumu i urinu, određivanje kompletne krvne slike, vrijednosti kalcija te bubrežnih parametara. Četvrtina bolesnika u promatranom periodu razvije multipli mijelom ili drugu B-staničnu neoplazmu. Najznačajniji prognostički čimbenici koji ukazuju na progresiju bolesti su vrijednosti M-komponete veće od 15 g/L te poremećen omjer lakih lanaca imunoglobulina (< 0,26 ili > 1,65) te hipogamaglobulinemija.
MULTIPLI MIJELOM
Definicija. Multipli mijelom predstavlja malignu bolest hematopoetskoga sustava obilježenu nekontroliranom klonalnom proliferacijom plazma stanica unutar koštane srži koje zadržavaju sposobnost sinteze i izlučivanja imunoglobulina (cijele molekule i/ili samo lakih lanaca), odnosno M-komponente. Osim M-komponente, karakteristična obilježja multiploga mijeloma uključuju osteolitičke promjene i koštanu destrukciju uz posljedičnu anemiju i hiperkalcijemiju te poremećaj bubrežne funkcije.
Epidemiologija. Multipli mijelom čini oko 1 % malignih oboljenja, a nakon ne-Hodgkinovih limfoma druga je najčešća hematopoetska maligna bolest s prosječnom godišnjom incidencijom od oko 4 do 5 novootkrivenih bolesnika na 100 000 stanovnika. Bolest se većinom javlja u starijoj životnoj dobi s najčešćom pojavnosti u šestom desetljeću života. Također, bolest se češće javlja kod muškaraca u odnosu na žene, a češća je kod osoba pripadnika crne rase.
Etiopatogeneza. Uzrok i mehanizam nastanka multiploga mijeloma nisu poznati. Kao rizični čimbenici za razvoj ove bolesti navode se genetska predispozicija (Tablica 6.39.), utjecaj ionizirajućega zračenja, kronična antigena stimulacija virusima te dugotrajna izloženost različitim kemikalijama kao što su pesticidi, herbicidi i sredstva za čišćenje. Danas se smatra da se inicijalni događaj u razvoju multiploga mijelom zbiva u koštanoj srži u kojoj dolazi do antigenskoga podržaja plazmablasta, odnosno B-limfocita koji su prošli proces diferenciranja u limfnom čvoru i migrirali u koštanu srž kako bi završili svoju diferencijaciju prema zrelim plazma stanicama. Nastanak kromosomskih promjena tijekom antigenske stimulacije plazmablasta može rezultirati stvaranjem poremećenih klonalnih stanica. Najčešće se radi o kromosomskim promjenama kromosoma 13 i 14 kao što su t(11;14), t(14;16), t(4;14) ili del(13) koje se mogu naći kod više od pola bolesnika. U toj fazi nastale klonalne stanice još nemaju svojstva malignih stanica te takvo stanje obično predstavlja monoklonalnu gamapatiju neodređenoga značenja. Stjecanjem dodatnih genetskih mutacija koje pogađaju tumor supresorske gene ili proonkogene (MYC, NRAS, KRAS, RB1, p53) stanice poprimaju obilježja malignih stanica što dovodi do karakterističnih obilježja bolesti (koštana destrukcija, infiltracija koštane srži, bubrežno oštećenje, poremećaj stvaranja imunoglobulina).
|
Tablica 6.39. Genetski rizični faktori
|
|
Visoki rizik
|
Srednji rizik
|
Standardan rizik
|
|
del(17p)
t(14;16)
t(14;20)
|
Medijan ukupnog preživljenja 3 godine
|
t(4;14)
del(13)
hipodiploidija
|
Medijan ukupnog preživljenja 4 – 5 godina
|
t(11;14)
t(6;14)
ostalo
|
Medijan ukupnog preživljenja 8 – 10 godina
|
| |
|
|
|
|
|
Nastanak osteolitičkih lezija. Jedno od osnovnih obilježja bolesti destrukcija je koštanoga tkiva kao posljedica prekomjerne aktivacije osteoklasta. Uslijed pojačanoga lučenja različitih medijatora iz tumorskih stanica dolazi do povećane ekspresije RANKL molekula na površini osteoblasta (a vjerojatno i samih tumorskih stanica) koje se vežu na svoje RANK receptore na površini osteoklasta, čime se oni aktiviraju i resorbiraju kost. Tomu pridonosi i smanjeno lučenje osteoprotegerina (OPG) koji inhibira tu RANKL-RANK interakciju. Interakcije RANKL, RANK i OPG detaljnije su opisane u poglavlju o poremećajima koštanoga metabolizma. Osim toga, i drugi medijatori, kao što su IL-3, IL-1B, IL6, DKK1 i dr., mogu imati stimulirajući učinak na osteoklaste, a inhibirajući na osteoblaste. Posljedično tim događajima dolazi do osteopenije, sklonosti patološkim frakturama te hiperkalcijemije.
Infiltracija koštane srži. Proliferacija malignoga klona plazma stanice unutar koštane srži rezultira potiskivanjem normalnoga hematopoetskoga tkiva uslijed čega se razvijaju anemija (normocitna, normokromna), neutropenija i trombocitopenija. Toj pancitopeniji pridonose i različiti medijatori koji se pojavljuju u takvom mikrookolišu i inhibirajuće djeluju na matične krvotvorne stanice.
Bubrežno oštećenje. Oštećenje bubrežne funkcije kod bolesnika s multiplim mjelomom (mijelomski bubreg, mijelomska nefropatija) posljedica je taloženja lakih lanaca u bubrežnim tubulima što dovodi do njihove degeneracije i atrofije te uslijed hiperkalcijemije i hiperkalciurije uvjetovane pojačanom koštanom razgradnjom. Mijelomska nefropatija klinički se prezentira postupnim razvojem kronične bolesti bubrega.
Poremećaj stvaranja imunoglobulina. Jedno od osnovnih obilježja tumorskih plazma stanica pojačana je produkcija M-komponente (cijeli i dijelovi imunoglobulina), što može biti udruženo s razvojem hiperviskoznoga sindroma, kao i stečene imunodeficijencije (proizvodnja monoklonskih imunglobulina dovodi do pareze normalnih imunoglobulina), što je obično praćeno učestalim respiratornim infekcijama. Kod manjega se broja bolesnika (< 3 %) ne može detektirati M-komponenta u serumu ili urinu, pa se takav oblik multiploga mijeloma naziva nesekretorni multipli mijelom.
Klinička slika. Kliničke manifestacije multiploga mijeloma posljedica su prethodno opisanih patofizioloških zbivanja. Najranije manifestacije na koje se bolesnici žale su umor, malaksalost, gubitak teka i tjelesne težine te bolovi u kostima, posebice gornjim i donjim ekstremitetima te kralježnici. Tada bolesnici često već imaju i patološke frakture, koje mogu biti uzrokom različitih neuroloških ispada (npr. kompresija na izlazišta živaca pri patološkim prijelomima kralježaka). Uslijed hiperkalcijemije se bolesnici mogu žaliti na konstipaciju i mučninu, poliuriju i polidipsiju, a mogu biti i konfuzni. Posljedično anemiji razvija se anemični sindrom, a trombocitopeniji sklonost krvarenju. Smanjena imunološka funkcija rezultira učestalim infekcijama, osobito inkapsuliranim bakterijskim sojevima. Bubrežno oštećenje razvija se postupno i u trenutku postavljanja dijagnoze najčešće se prezentira asimptomatskim patološkim nalazima krvi i urina. Pojava hiperviskoznoga sindroma je rijetka, a može se manifestirati epistaksom, krvarenjem iz usne šupljine, purpurom, poremećajem vida, glavoboljom, otežanim disanjem i bolovima u prsima. Učestalost kliničkih manifestacija i patoloških nalaza u trenutku postavljanja dijagnoze prikazana je u Tablici 6.40.
|
Tablica 6.40. Učestalost kliničko-laboratorijskih nalaza u trenu postavljanja dijagnoze multiploga mijeloma
|
|
Kliničko-laboratorijski nalaz
|
Učestalost pri dijagnozi
|
|
Koštane lezije (bolovi, frakture, radiološki nalaz)
|
80 %
|
|
Anemija (hemoglobin < 120 g/L)
|
75 %
|
|
Oštećenje bubrežne funkcije (kreatinin > 2 mg/dL)
|
20 %
|
|
Hiperkalcijemija (serumski kalcij > 11 mg/dL)
|
15 %
|
|
Prisutnost M-komponentne (elektroforeza ili imunofiksacija serumskih proteina)
|
97 %
|
|
Prisutnost klonalnih plazma stanica u koštanoj srži (> 10%)
|
96 %
|
Dijagnoza multiploga mijeloma temelji se na laboratorijskim nalazima, nalazima biopsije koštane srži te radiografskoj dijagnostici. Dodatno se provode i druge dijagnostičke metode s ciljem otkrivanja proširenosti bolesti i zahvaćanja drugih tkiva i organa. Dijagnostički kriteriji za multipli mijelom prikazani su u Tablici 6.41.
Laboratorijski nalazi. Hematološki krvni nalazi najčešće upućuju na normocitnu i normokromnu anemiju, dok leukopeniju (neutropeniju) i trombocitopeniju nalazimo u uznapredovalim fazama bolesti. Biokemijski nalazi obično upućuju na hiperkalcijemiju, a ovisno o stupnju bubrežnoga oštećenja mogu se naći i povišene vrijednosti ureje i kreatinina. Sedimentacija eritrocita je ubrzana, a ukupni proteini su uredni ili povišeni. Ukoliko se na temelju kliničke slike i ovih osnovnih laboratorijskih nalaza posumnja na multipli mijelom, indicira se proširena obrada koja uključuje: elektroforezu serumskih proteina, kvantitativno određivanje vrijednosti imunoglobulina (IgG, IgA, IgM, IgD), imunofiksaciju proteina seruma i urina, određivanje slobodnih lakih lanaca u serumu i urinu te njihovoga omjera (kappa/lambda omjer) i određivanje koncentracije β2-mikroglobulina u serumu.
Otkrivanje M-komponente omogućuju metode elektroforeze ili imunofiksacije proteina seruma i/ili urina (danas se preporuča korištenje metode imunofiksacije s obzirom na njezinu veću osjetljivost i specifičnost u odnosu na elektroforezu), što čini ključni dijagnostički nalaz. M-komponenta pojavljuje se kao šiljak u određenom dijelu elektroforetske krivulje, i to najčešće u području gama i beta dijela. Kod bolesnika sa sumnjom na multipli mijelom kod kojega se spomenutim metodama ne otkriva M-komponenta moguće je i određivanje slobodnih lakih lanaca u serumu (i/ili urinu) što omogućuje detekciju multiploga mijeloma karakteriziranoga lučenjem samo lakih lanaca imunoglobulina (mijelom lakih lanaca), a smatra se da se to susreće kod oko dvije trećine bolesnika koji imaju neskretorni oblik bolesti. Također, njihov omjer (kappa/lambda omjer) ima važno prognostičko značenje. Koncentracija β-2-mikroglobulina odražava ukupnu tumorsku masu zbog čega ima velik dijagnostičko-prognostički značaj. Kvantitativno određivanje vrijednosti imunoglobulina (IgG, IgA, IgM, IgD) daje uvid u prirodu i veličinu M-komponente, kao i stupanj imunopareze ostalih (nemijelomskih) imunoglobulina. Najčešće M-komponentu takvih bolesnika čine imunoglobulin G (52 %), imunoglobulin A (21 %), laki lanci ili Bence-Jonesova proteinemija (16 %) te imunoglobulin D (2 %).
Biopsija koštane srži. Tipičan nalaz uzorka koštane srži ukazuje na prisutnost klonalnih plazma stanica čiji ukupni udio prelazi 10 %, kod manje od 5 % bolesnika njihov udio je manji od 10 %. Stanice malignoga klona na svojoj površini obično eksprimiraju molekule CD38, CD138, CD56 i CD19 što se može dokazati imunofenotipizacijom. Klonalnost plazma stanica određuje se omjerom kappa/lambda lanaca (omjer veći od 4:1 ukazuje na klonalnost populacije plazma stanica). Citogenetskim i molekularnim metodama mogu se dokazati opisane kromosomske i genetske promjene.
Radiološka dijagnostika. Radiološkim dijagnostičkim metodama dokazuje se prisutnost osteolitičkih lezija i/ili patoloških fraktura. Pri tome se najčešće koristi klasični rendgenogram cijeloga skeleta (kraniogram, kralježnica, prsni koš, nadlaktica i natkoljenica). Magnetska rezonanca preporuča se kod bolesnika s laboratorijskim nalazima suspektnim na multipli mijelom s urednim klasičnim radiogramom. U tu se svrhu može koristiti i kompjuterizirana tomografija.
|
Tablica 6.41. Dijagnostički kriteriji multiploga mijeloma
|
|
Asimptomatski multipli mijelom
|
Simptomatski (aktivan) multipli mijelom
|
|
Monoklonski protein prisutan u serumu (IgG ≥ 30 g/L; IgA > 30 g/L) ili u urinu (≥ 500 mg/24 sata) i/ili infiltracija koštane srži klonskim plazma stanicama 10% do 60%;
Nema oštećenja drugih organa mijelomom ili postojanja drugih znakova mijeloma ili amiloidoze;
Bence-Jones protein >500 g/24 sata.
|
Klonalne plazma stanice u koštanoj srži ≥ 10% uz koštani ili ekstramedularni plazmacitom (dokazan biopsijom) uz barem jedno od sljedećih obilježja:
1)CRAB obilježja:
Koncentracija kalcija iznad 2.75 mmol/L;
Bubrežna insuficijencija ili KEK < 40ml/min ili serumski kreatinin > 177 umol/L;
Anemija, hemoglobin < 100 g/L ili 20 g/L ispod normale;
Jedna ili više osteolitičkih lezija dokazanih RTG-om, CT-om ili PET- CT.
2)Biomarkeri maligniteta:
Jedna ili više fokalnih lezija na MRI > 5 mm;
≥ 60% klonalnih stanica u koštanoj srži;
Odnos zahvaćenih i nezahvaćenih slobodnih lakih lanaca u serumu: ≥ 100 uključujući kappa ili < 0.01 uključujući lambda.
|
Diferencijalna dijagnoza. Mulipli mijelom i njegove varijante (kasnije opisano) treba razgraničiti od drugih limfoproliferativnih bolesti, kao što su kronična limfocitna leukemija, Waldenstromova makroglobulinemija, ne-Hodgkinovi limfomi te krioglobulinemija. Također je multipli mijelom i njegove varijante potrebno razlikovati i od reaktivne (benigne) poliklonalne hipergamaglobulinemije koju možemo vidjeti u bolesnika s kroničnom upalom ili cirozom.
Liječenje bolesnika s multiplim mijelom temelji se na dobi i kondicijskom stanju bolesnika, odnosno ovisi tome jesu li bolesnici kandidati za transplantacijsko liječenje.
Bolesnici koji su kandidati za transplantacijsko liječenje (bolesnici mlađi od 65 godina, dobroga općeg stanja i bez značajnijih komorbiditeta) liječe se intenziviranom kemoterapijom nakon koje slijedi autologna transplantacija krvotvornih matičnih stanica. Kod većine bolesnika inicijalnu kemoterapiju čini imunomodulatorni lijek (lenalidomid), inhibitor proteasoma (bortezomib) uz umjerene ili visoke doze deksametazona. Kod bolesnika koji ne pokazuju odgovor u obzir dolazi i primjena drugih lijekova kao što su pomalidomid (imunomodulator), daratumumab (anti-CD38 moonoklonalno antitijelo), elotuzumab (anti-SLAMF7 monoklonalno antitijelo), novi inhibitori proteasoma (karfilzomib ili iksazomib) ili panobinostat (inhibitor histonske deacitilaze). Nakon inicijalnoga liječenja primjenjuje se autologna transplantacija krvotvornih matičnih stanica uz primjenu visokih doza melfalana, alkilirajućega kemoterapeutika. Kod bolesnika kod kojih se ne postigne dobar posttransplantacijski odgovor u obzir dolazi i druga, tzv. tandem-transplantacija. Nakon postignutoga odgovora (redukcija M-komponente za više od 90 %) može se nastaviti terapija održavanja s ciljem produženja remisije, a u tu se svrhu mogu koristiti lenalidomid ili talidomid, koji nose rizik od sekundarnih malignih oboljenja, ili inhibitori proteasoma. Terapija održavanja osobito se preporuča za visoko rizične bolesnike.
Bolesnici koji nisu kandidati za transplantacijsko liječenje (bolesnici stariji od 65 godina, lošijega općeg stanja s ili bez značajnijih komorbiditeta) imaju terapiju sastavljenu od peroralne primjene melfalana i prednizolona uz dodatak imunomodulatora (talidomid) ili inhibitora proteasoma (bortezomib) uz provođenje simptomatskoga liječenja.
Simptomatsko liječenje i liječenje komplikacija. Bifosfonati (pamidronat, zoledronat) predstavljaju osnovu simptomatskoga liječenja bolesnika s multiplim mijelomom s obzirom na njihov povoljan antiresorptivan učinak. Njihova primjena dokazano smanjuje bolove u kostima te smanjuje rizik od nastanka patoloških fraktura, a koristi se i u liječenju hiperkalcijemije. Njihovu dugotrajnu primjenu ograničava mogućnost nastanka osteonekroze donje čeljusti i drugih koštanih regija. U slučaju pojave patoloških fraktura praćenih neurološkim ispadima u obzir dolazi radioterapija zahvaćenoga područja i/ili kirurška intervencija. Liječenje mijelomske nefropatije sastoji se, uz specifično liječenje kojime se smanjuje sekrecija M-komponente, od održavanja zadovoljavajuće diureze što se uz hidraciju može postići i dodatkom diuretika. U specifičnim situacijama, kao što je naglo pogoršanje bubrežne funkcije uz značajno povišene vrijednosti lakih lanaca imunoglobulina u serumu, u obzir dolazi i plazmafereza s ciljem njihova uklanjanja i sprečavanja daljnjega taloženja u tubulima. U uznapredovaloj fazi bolesti indicirano je liječenje hemodijalizom. Pored navedenih mjera potrebno je korigirati anemiju, prevenirati i na vrijeme liječiti infekcije (kod svih bolesnika indicirano je cijepljenje protiv pneumokoka s obzirom na to da su ti bolesnici skloni infekcijama inkapsuliranim sojevima) te izbjegavati primjenu nesteroidnih protuupalnih lijekova zbog negativnoga učinka na bubrežnu funkciju.
Prognoza. Uz primjenu novih lijekova prognoza bolesnika s multiplim mijelom znatno je napredovala u zadnjih desetak godina tako da danas medijan preživljenja iznosi više od 7 godina. Durie-Salmon klasifikacija odražava ukupnu količinu tumora i u vezi je s procjenom količine tumorskih stanica (6.43.). Internacionalni sustav stupnjevanja (ISS) je prognostička klasifikacija koja ima veće prognostičko značenje kombinacijom dvaju parametara koji ukazuju na prognozu, a to su β2-mikroglobulin i albumin. Danas postji i njezina revidirana verzija kojoj su uz prijašnje kriterije dodali citogenetske promjene i vrjednosz LDH (Tablica 6.42.). Preporuča se određivanje stadija bolesti prema obje klasifikacije pri dijagnozi, a što određenu vrijednost ima i pri procjeni stanja u kasnijim fazama bolesti. Prognoza ovih bolesnika usko je vezana i s genetskim abnormalnostima koje nalazimo u tumorskim stanicama kako je to i navedeno u Tablici 6.39.
|
Tablica 6.42. Prognostička R-ISS klasifikacija
|
|
Parametar/preživljenje
|
Prvi stadij
|
Drugi stadij
|
Treći stadij
|
|
β2-mikroglobulin
|
< 3,5 g/mL
|
< 3,5 ili 3,5 - 5,5 g/mL
|
> 5,5 g/mL
|
|
Serumski albumin
|
> 5 g/L
|
|
Bez obzira
|
|
FISH
|
Standardni rizik i
|
NIJE R-ISS I ILI R-ISS III
|
Visoki rizik ili
|
|
LDH
|
Uredna vrijednost
|
Povišen
|
|
Preživljenje bez progresije
|
66 mjeseci
|
42 mjeseci
|
29 mjeseci
|
|
Tablica 6.43. Durie-Salmon klasifikacija proširenosti bolesti
|
|
Stadij I (mala stanična masa)
|
Stadij II (srednja stanična masa)
|
Stadij III (velika stanična masa)
|
|
Prisutno sve od sljedećeg:
Hemoglobin > 100g/L;
Kalcij u serumu normalan ili tek blago povišen;
Normalan nalaz rtg skeleta ili solitarni plazmocitom kosti;
Niska produkcija M komponente (IgG < 50 g/L, IgA < 30 g/L);
M komponenta lakih lanaca u urinu elektroforezom < 4g/24 h.
|
Ne uklapa se niti u stadij I niti u stadij II bolesti.
|
Prisutno jedno ili više od sljedećeg:
Hemoglobin
Kalcij u serumu značajno povišen;
Uznapredovale litičke lezije kostiju;
Visoka produkcija M komponente (IgG >70 g/L, IgA > 50 g/L);
M komponenta lakih lanaca u urinu elektroforezom > 12 g/24h.
|
Ostali klinički oblici povezani s multiplim mijelomom su asimptomatski multipli mijelom, plazmastanična leukemija, nesekretorni mijelom, osteosklerotski mijelom (POEMS sindrom), solitarni koštani plazmacitom te ekstramedularni plazmacitom.
Asimptomatski (šuljajući, engl. „smoldering“) multipli mijelom multipli mijelom obilježava prisutnost M-komponente u serumu (> 3 g/dL) ili prisutnost 10 do 60 % klonalnih plazma stanica u koštanoj srži, uz odsutnost ostalih karakterističnih kliničkih obilježja multiploga mijeloma. Biološka obilježja ovoga stanja slična su onome kod monoklonalne gamapatije neodređenoga značenja, ali sa znatno višim rizikom od progresije u simptomatsku bolest. Specifično liječenje nije potrebno, osim kod bolesnika s vrlo visokim rizikom, a to su bolesnici s omjerom slobodnih lakih lanaca u serumu većim od 100 ili s prisutnošću jedne ili više radiološki detektibilne osteolitičke lezije. Njihovo liječenje jednako je opisanome liječenju bolesnika sa simptomatskim multiplim mijelomom. Bolesnike koje se ne liječi potrebno je češće kontrolirati, u pravilu svaka tri do četiri mjeseca kako bi se na vrijeme utvrdila eventualna progresija bolesti.
Plazmastanična leukemija definira se prisutnošću više od 20 % plazma stanica u perifernoj krvi ili je apsolutni broj plazma stanica u perifernoj krvi veći od 2 x 109/L. Ona može biti primarna i sekundarna. O primarnoj govorimo kada se bolest dijagnosticira u leukemijskoj fazi bolesti, a o sekundarnoj kada ona nastaje iz ranije poznatoga multiplog mijeloma. Karakteristike primarne plazmastanične leukemije su mlađa životna dob u trenutku pojavnosti, veća učestalost hepatosplenomegalije te manje izražena M-komponenta u serumu. Liječenje podrazumijeva agresivnu kemoterapiju koja uključuje različite kemoterapeutike (bortezomib, talidomid, cisplatina, doksorubicin, ciklofosfamid, etopozid) i deksametazon u kombinaciji s autolognom transplantacijom koštane srži i terapijom održavanja koja se temelji na inhibitorima proteasoma (bortezomib). Prognoza bolesnika sa sekundarnom plazmastaničnom leukemijom lošija je u odnosu na primarnu.
Nesekretorni multipli mijelom obilježen je izostankom M-komponente u serumu, što je slučaj kod oko 3 % bolesnika. Dijagnoza se kod takvih bolesnika temelji isključivo na nalazu biopsije koštane srži. Liječenje je jednako kao i kod simptomatskoga, tipičnoga multiplog mijeloma.
Osteosklerotski mijelom (POEMS sindrom) označava kliničko stanje koje obilježavaju upalno-demijelinizirajuća polineuropatija, organomegalija, endokrinopatija, prisutnost M-komponente u serumu te promjene na koži uz prisutnost sklerotičnih koštanih lezija. Točan uzrok i mehanizam nastanka toga poremećaja nisu poznati. U sklerotičnim koštanim lezijama mogu se naći klonalne plazma stanice, a prisutnost hiperkalcijemije i bubrežne insuficijencije je rijetka. Dijagnoza se potvrđuje nalazom biopsije koštane srži, a liječenje se provodi zračenjem ako se radi o ograničenim (lokaliziranim) sklerotičnim lezijama, dok se kod diseminirane bolesti primjenjuje liječenje kao i kod klasičnoga multiplog mijeloma (kemoterapija, transplantacija).
Solitarni koštani plazmacitom predstavlja tumorsku koštanu leziju koja se sastoji od monoklonalnih plazma stanica identičnih onima kakve nalazimo kod bolesnika s multiplim mijelomom. Za postavljanje dijagnoze solitarnoga plazmacitoma potrebno je radiološkim metodama isključiti postojanje drugih sličnih lezija, kao i prisutnost tumorskih plazma stanica u koštanoj srži. M-komponenta može, ali i ne mora biti prisutna u serumu u trenutku postavljanja dijagnoze, a njezina pojava nakon terapije ukazuje na povišen rizik od progresije u multipli mijelom. Temelj liječenja predstavlja radioterapija, a do sada nema dokaza da primjena adjuvantne kemoterapije smanjuje rizik od progresije u multipli mijelom. Progresija u multipli mijelom obično se zbiva unutar tri godine od postavljanja dijagnoze. Ukupno preživljenje ovih bolesnika duže je od deset godina.
Ekstramedularni plazmacitom predstavlja tumorsko tkivo građeno od monoklonalnih plazma stanica, a pojavljuje se izvan koštane srži, najčešće u gornjim dišnim putevima (nos, sinusi, nazofarinks, larinks), a može se pojaviti i u probavnom, središnjem živčanom sustavu, mokraćnom mjehuru, dojci ili limfnim čvorovima. Dijagnoza se postavlja na temelju histološkoga pregleda uzorka tumorskog tkiva te isključenjem prisutnosti tumorskih plazma stanica u koštanoj srži. Liječenje se temelji na kirurškom odstranjenju tumora uz naknadnu radioterapiju. Ekstamedularni plazmacitomi skloni su recidiviranju, a mogu progredirati i u klasični multipli mijelom.
WALDENSTROMOVA MAKROGLOBULINEMIJA
Definicija i epidemiologija. Waldenstromova makroglobulinemija ili primarna makroglobulinemija nastaje kao posljedica nekontrolirane proliferacije limfocita i/ili plazma stanica koje stvaraju monoklonalni IgM protein. Radi se o rijetkom oboljenju koje se obično pojavljuje u starijoj životnoj dobi (najčešće u petom i šestom desetljeću) s nešto češćom pojavnošću kod muškaraca u odnosu na žene.
Etiopatogeneza. Točan uzrok i mehanizam razvoja bolesti, kao i rizični čimbenici, nisu poznati. S obzirom na to da se kod većine tih bolesnika nalazi mutacija MYD88 gena, smatra se da on može biti u nekoj mjeri odgovoran za nastanak klonalne proliferacije limfocita ili plazma stanica.
Klinička slika. Većina bolesnika ima dug asimptomatski period. Prvi znakovi bolesti obično podrazumijevaju osjećaj umora i malaksalosti te znakove anemije. U kasnijim fazama bolesti mogu se pojaviti znakovi hiperviskoznoga sindroma uslijed pretjeranoga izlučivanja IgM-a (glavobolja, zamućen vid, poremećaj stanja svijesti, dispneja, stenokardija) te sklonost krvarenju zbog interferencije IgM-a s faktorima koagulacije. Pojava koštanih litičkih lezija, hiperkalcijemije i renalne insuficijencije je rijetka. U većine bolesnika može se uočiti limfadenopatija i hepatosplenomegalija.
Dijagnoza se postavlja na temelju laboratorijskih nalaza te nalaza biopsije/punkcije koštane srži. Temeljni laboratorijski nalaz podrazumijeva elektroforezu serumskih proteina koja ukazuje na prisutnost serumskoga monoklonalnog imunoglobulina M, čije su vrijednosti obično više od 15 g/L, dok u koštanoj srži nalazimo više od 10 % limfoblastičnih stanica koje na svojoj površini eksprimiraju CD19, CD20 i CD22 antigene.
Liječenje i prognoza. Asimptomatski bolesnici ne zahtijevaju specifično liječenje, nego samo praćenje. Simptomatsko liječenje podrazumijeva liječenje hipervisokoznoga sindroma koje se obično temelji na pojačanoj hidraciji, a u najtežim slučajevima u obzir dolazi i plazmafereza. Osnovu specifičnoga liječenja čini primjena rituksimaba kao samostalnoga lijeka ili u kombinaciji s ciklofosfamidom i deksametazonom (RCD protokol) ili bendamustinom (RB protokol). Kod bolesnika s pozitivnom MYD88 mutacijom u slučaju refrakternosti ili relapsa u obzir dolazi primjena ibrutiniba (BTK inhibitor). Autologna transplantacija krvotvornih stanica indicirana je kod bolesnika s refrakternom ili recidivirajućom bolesti. Waldenstromova makroglobulinemija je načelno bolest indolentna tijeka s medijanom preživljenja od oko pet godina, iako više od 10 % bolesnika živi duže od 15 godina.
BOLEST TEŠKIH LANACA
Definicija. Bolest teških lanaca podrazumijeva plazmastanične neoplazme čije stanice stvaraju i izlučuju teške lance imunoglobulina koji čine M-komponentu u serumu, a ovisno o vrsti teških lanaca koje luče razlikujemo bolest gama (γ) teških lanaca, bolest alfa (α) teških lanaca te bolest mi (µ) teških lanaca.
Bolest alfa (α) teških lanaca (Seligmannova bolest) najčešći je oblik bolesti teških lanaca, a karakterizira ju pojačano stvaranje i lučenje alfa teškog imunoglobulinskog lanaca koji čini M-komponentnu, što se dokazuje metodom imunofiksacije. Javlja se u mlađoj životnoj dobi i to najčešće na geografskom području Mediterana i Srednjega istoka. Bolest najčešće zahvaća probavni sustav pri čemu tumorske stanice infiltriraju jejunum te dovode do sindroma malapsorpcije. Bolest se rijetko prezentira zahvaćanjem dišnoga sustava. Inicijalno liječenje bolesnika sastoji se od primjene antimikrobnih lijekova (tetraciklini), a kod bolesnika koji ne pokazuju odgovor indicirana je kemoterapija slična onoj za liječenje ne-Hodgkinovih limfoma (CHOP protokol).
Bolest gama (γ) teških lanaca (Franklinova bolest) karakterizirana je nalazom monoklonalnoga teškog gama lanca koji čini M-komponentu na temelju metode imunofiksacije. Bolest može imati varijabilan tijek, od asimptomatskoga i indolentnog do agresivnoga i brzoprogresivnog. Liječenje se provodi samo kod simptomatskih bolesnika, a temelji se na melfalanu i prednizolonu ili protokolima koji se koriste u liječenju ne-Hodgkinova limfoma (CHOP protokol). Prognoza je varijabilna.
Bolest mi (µ) teških lanaca najrjeđi je oblik bolesti teških lanaca. Karakterizira ju povećano stvaranje i izlučivanje mi teških imunoglobulinskih lanaca, što se dokazuje metodom imunofiksacije. Najraširenija je u području Afrike i povezuje se s parazitarnim crijevnim infestacijama. Klinički tijek bolest je nepredvidiv, a preživljenje se kreće od svega nekoliko mjeseci do više godina. Liječenje se temelji na alkilirajućim kemoterapeuticima i kortikosteroidima.
FIZIOLOGIJA KOAGULACIJSKOG SUSTAVA
Koagulacijski sustav je kompleksan sustav čiji je cilj stvaranje krvnoga ugruška u procesu hemostaze, odnosno procesu zaustavljanja krvarenja. Reguliran je brojnim interakcijama između stanica i različitih serumskih proteina. Glavne komponente koagulacijskoga sustava su trombociti, endotelne stanice te koagulacijski faktori. Krajnji rezultat jednom aktiviranoga koagulacijskog sustava je nastanak fibrinske mreže koja zajedno s nakupljenim trombocitima čini krvi ugrušak.
Trombociti, krvne pločice, temeljni su dio koagulacijskoga sustava. Njihova membrana važan je izvor fosfolipida potrebnih za normalnu funkciju koagulacijskih faktora, a osim toga sadrže i različite receptore koji omogućuju njihovo vezanje za endotelne stanice (adhezija), kao i međusobno povezivanje (agregacija), što ima bitnu ulogu u procesu nastanka krvnoga ugruška. Da bi trombociti sudjelovali u procesu hemostaze, moraju se najprije aktivirati. Pri aktivaciji trombocita dolazi do promjene oblika trombocita, promjene ekspresije i strukture specifičnih trombocitnih receptora, kao i sekrecije različitih prokoagulantnih molekula. Tu aktivaciju potiču induktori preko unutarstaničnih signalnih puteva, a najvažniji su tromboksan A2, adenozin-difosfat, kolagen te trombin. Promjena oblika trombocita označava njihovo izduživanje čime se povećava kontaktna površina između trombocita i endotela. Pojačava se i mijenja funkcija specifičnih trombocitnih receptora od kojih je najbitniji integrin GPIIb-IIIa koji omogućava stvaranje mostova između trombocita, čime potiče njihovu međusobnu agregaciju (nakupljanje). Povezivanje trombocita s kolagenom stijenke krvnih žila nakon oštećenja endotela omogućava von Wilenbrandov faktor. Sekretorni odgovor trombocita nakon njihove aktivacije podrazumijeva degranulaciju i oslobađanje različitih biološki aktivnih molekula, kao što su aktivatori trombocita (tromboksan A2, adenozin-difasfat), vazokonstriktori (serotinin, gvanozin-difosfat) te kemokini (npr. IL-8) koji izazivaju kemotaksiju neutrofila i monocita. Osim toga, anionski fosfolipidi koji se u neaktivnim trombocitima nalaze s unutarnje strane membrane trombocita prilikom aktivacije prelaze na vanjsku stranu te čine podlogu na kojoj se odvija aktivacija koagulacijskih faktora.
Endotelne stanice također imaju veliku ulogu u procesu hemostaze. U normalnim uvjetima one posjeduju mehanizme kojima sprečavaju adheziju, aktivaciju i agregaciju trombocita čime se sprečava nastanak krvnoga ugruška unutar neoštećenih krvnih žila. Navedeno je posljedica djelovanja elektrostatskih sila, ali i sinteze određenih bioloških molekula među kojima je najvažniji enzim ADP-aza koji razgrađuje trombocitni aktivator adenozin-difosfat. Također, endotelne stanice posjeduju i faktor aktivacije trombocita (PAF) koji se oslobađa prilikom njihova oštećenja, a on potiče aktivaciju trombocita.
Koagulacijski faktori omogućavaju stvaranje fibrinske mreže koja omata nakupljene trombocite stvarajući pravi krvni ugrušak. Oni se u cirkulaciji kreću u neaktivnom obliku, a većina njih djeluje kao enzimi (serinske proteaze) koji imaju sposobnost međusobne aktivacije, tzv. amplifikacijski mehanizam. Faktori se označavaju rimskim brojem bez oznake „a“ kada su u inaktivnom obliku (npr. FII ili protrombin) te oznakom „a“ ako su u aktivnom obliku (npr. FIIa ili trombin). Većina ih se sintetizira u jetri, osim faktora XIII koji je trombocitnoga porijekla te faktora VIII kojega stvaraju endotelne stanice. Sinteza faktora II, VII, IX i X ovisi o aktivnosti enzima gama-karboksilaze u jetri, za čiju je normalnu aktivnost potreban vitamin K.
Hemostaza je naziv za proces zaustavljanja krvarenja iz oštećene krvne žile, a može se podijeliti u tri zasebna procesa: 1) primarna hemostaza, 2) sekundarna hemostaza te 3) fibrinoliza.
Primarna hemostaza predstavlja rani odgovor na endotelno oštećenje koji podrazumijeva vazokonstrikciju posredovanu simpatikusom (s ciljem smanjenja protoka krvi kroz oštećeno područje), kao i aktivaciju trombocita. Izlaganje trombocita kolagenu ili trombinu dovodi do njihove aktivacije koja, kako je navedeno, rezultira promjenom oblika te ekspresijom molekula na svojoj površini, a prateće povećanje unutarstanične koncentracije kalcija vodi prema degranulaciji i oslobađanju bioloških aktivnih molekula koje dalje promoviraju koagulacijski sustav i vode prema stvaranju tromba.
Sekundarna hemostaza podrazumijeva aktivaciju i međusobnu interakciju koagulacijskih faktora s konačnim rezultatom formiranja fibrinske mreže. Klasično se proces stvaranja fibrinske mreže opisuje kroz dva puta: unutarnji (intrizični) i vanjski (ekstrizični) koji se potom ujedinjuju u zajednički put.
Zajednički put započinje aktivacijom FX u FXa koji aktivira protrombinazu, a ona pretvara protrombin (FII) u aktivirani oblik trombin (FIIa), koji potom pretvara fibrinogen (FI) u fibrin (FIa). Tako nastaje fibrinska mreža koja zajedno s agregiranim trombocitima, ali i drugim krvnim stanicama, čini tromb. Fibrinska mreža nastaje udruživanjem fibrina u fibrinske polimere koji se međusobno povezuju što je posredovano aktiviranim faktorom XIII (FXIIIa).
Vanjski put započinje ozljedom endotela i otpuštanjem tkivnoga faktora (TF) koji se još naziva i tromboplastin. On aktivira FVII te tako nastaje FVIIa. Kompleks TF-FVIIa potom aktivira FX u FXa kojim započinje zajednički put zgrušavanja. Fxa, osim što sudjeluje u direktnoj aktivaciji protrombinaze, sudjeluje i u aktivaciji FV u Fva, koji također potpomaže aktivaciju protrombinaze, te tako i neizravno potpomaže njezinu aktivaciju, što je također dio vanjskoga puta zgrušavanja.
Unutarnji put također započinje oštećenjem endotela, tj. izlaganjem krvi kolagenu koji se nalazi u stijenci krvne žile. Prvi korak toga puta aktivacija je FXII (Hagemanov faktor) pri čemu nastaje FXIIa koji djeluje na FXI, pri čemu nastaje FXIa. FXIa aktivira FIX te tako nastaje FIXa. Faktor IXa zajedno s FVIIIa (FVIII prethodno aktivira trombin), kalcijem i fosfolipidima na površini trombocita sudjeluje u aktivaciji FX u FXa.
Iako se ta dva puta odvojeno opisuju, oni su međusobno povezani i nadopunjuju se. Nakon ozljede se vanjski put puno brže aktivira (oko 15 sekundi), za razliku od unutarnjega puta kojemu treba nešto više vremena (nekoliko minuta). U cijelome procesu bitnu ulogu ima i kalcij koji djeluje kao katalizator većine navedenih reakcija te u njegovom odsustvu navedene reakcije ne bi niti bile moguće.
Fibrinoliza. U konačnici, dio koagulacijskog sustava čini i proces fibrinolize koji podrazumijeva razgradnju stvorene fibrinske mreže i tromba u cijelosti. Glavni fibrinolitički enzim je plazmin koji razgrađuje fibrinsku mrežu stvarajući degradacijske produkte koji se nazivaju D-dimeri. On nastaje aktivacijom iz plazminogena, a pretvorbu plazminogena u plazmin katalizira tkivni aktivator plazminogena (t-PA).
Osim fibrinolitičkoga puta, procesu koagulacije suprotstavljaju se i različite druge molekule koje pripadaju tzv. antikoagulacijskom sustavu, kao što su antitrombin (inhibira faktore II, IX, X, XI i XII), protein C (inhibira faktore V i VIII) te protein S (kofaktor aktiviranog proteina C). Nedostaci ovih antikoagulantnih molekula rezultirat će povećanom sklonosti zgrušavanju krvi.
SKLONOST KRVARENJU ILI HEMORAGIJSKA DIJATEZA
Uvod. Hemoragijska dijateza podrazumijeva klinički pojam kojime se opisuje sklonost bolesnika krvarenju, a može nastati uslijed različitih nasljednih ili stečenih poremećaja koagulacijskoga sustava. Nasljedni poremećaji koji dovode do hemoragijske dijateze su rjeđi, u pravilu označavaju poremećaj jednoga od faktora koagulacije, dok su stečeni poremećaji češći, a rezultiraju zahvaćanjem više koagulacijskih faktora.
NASLJEDNI POREMEĆAJI
Najčešći nasljedni poremećaji koji se prezentiraju hemoragijskom dijatezom su hemofilija A, hemofilija B te von Willenbrandova bolest.
HEMOFILIJA A
Definicija. Hemofilija A označava nasljednu sklonost krvarenju koja se javlja kao posljedica nedostatka ili smanjenje funkcije koagulacijskoga faktora VIII. Radi o koagulacijskom faktoru koji se u najvećoj mjeri stvara i luči iz endotelnih stanica, a krvotokom se kreće vezan za von Willenbrandov faktor (vWF) čime se sprečava njegova prerana razgradnja. Faktor VIII dio je unutarnjega puta zgrušavanja kojega aktivira trombin i zajedno s aktiviranim faktorom IX (FIXa), kalcijem i fosfolipidima sudjeluje u aktivaciji faktora X što čini inicijalni korak u zajedničkom putu zgrušavanja koji u konačnici rezultira stvaranjem fibrinske mreže i ugruška. Učestalost hemofilije A procjenjuje se na 1 na 10 000 muške djece (s obzirom na to da se radi o X-vezanom nasljeđivanju od te bolesti oboljeti mogu samo muške osobe, dok su žene zdravi prenosioci).
Etiopatogeneza i genetika. Hemofilija A nastaje kao rezultat nasljedne (rjeđe de novo) mutacije gena za faktor VIII koji se nalazi na dugom kraku X kromosoma (Xq28). Do danas je poznato i opisano više stotina mutacija koje mogu rezultirati apsolutnim ili smanjenim izostankom stvaranja faktora VIII ili stvaranjem afunkcionalnih oblika faktora VIII. S obzirom na to da se radi o recesivnom X-vezanom nasljeđivanju, vrijede uobičajena pravila nasljeđivanja ove bolesti: 1) gotovo sve oboljele osobe muškoga su spola (kod žena je pojava hemofilije A vezana uz druge kromosomske poremećaje kao što je Turnerov sindrom i sl.); 2) oboljeli muškarac nikad ne prenosi bolest na svoje sinove; 3) sve kćeri oboljeloga muškaraca su nosioci bolesti; 4) žene-nosioci bolest prenose na polovicu svojih sinova. Kod manje od 5 % bolesnika ne mogu se dokazati mutacije ovoga gena te kod njih etiologija toga poremećaja ostaje nepoznata. Posljedično manjku ili nefunkcionalnosti faktora VIII dolazi do poremećaja aktivacije zajedničkoga puta koagulacije (aktivacija faktora X) što rezultira sklonošću prema krvarenju.
Klinička slika. ežina kliničke slike ponajprije ovisi o vrsti mutacije i posljedičnoj aktivnosti faktora VIII. Bolest u prvome redu karakteriziraju spontana krvarenja ili krvarenja izazvana minimalnom traumom, i to najčešće u koži, mišićima i zglobovima. Krvarenje u zglob (hemartros) najkarakterističniji je znak kod takvih bolesnika. Iako se može pojaviti u bilo kojem zglobu, najčešće se viđa u koljenskom, lakatnom, gležanjskom i ramenom zglobu. Naknadna resorpcija i vezivna organizacija hematoma može dovesti do degenerativnih promjena i kontraktura (hemofilična artroptija). Krvarenja u druge sustave (hematurija, melena, hematemeza, subarahnoidalna krvarenja i sl.) relativno su rijetka i viđaju se pri najtežim oblicima bolesti. Ovisno o stupnju manjkavosti faktora VIII, razlikujemo tri stupnja bolesti (teška, umjerena i blaga hemofilija – Tablica 6.44.).
|
Tablica 6.44. Podjela hemofilije A prema težini
|
|
Oblik i učestalost
|
Vrijednost FVIII
|
Kliničke karakteristike
|
|
Blaga hemofilija (15 %)
|
> 5 IU/dL
|
Sekundarna krvarenja povezana s većim zahvatima ili traumama; obično se otkriva u starijoj dobi
|
|
Umjerena hemofilija (15 %)
|
1 – 5 IU/dL
|
Sekundarna krvarenja koja se javljaju pri manjim zahvatima i lakšim traumama
|
|
Teška hemofilija (70 %)
|
< 1 IU/dL
|
Spontana krvarenja koja se javljaju od djetinjstva, kada se ne liječi na odgovarajući način česte su kontrakture
|
Dijagnoza se postavlja na osnovi kliničke slike, laboratorijskih nalaza i genetskoga testiranja. U laboratorijskim je nalazima produljeno aktivirano parcijalno tromboplastinsko vrijeme (aPTV), kao odraz manjkavosti unutarnjega puta zgrušavanja, uredno protrombinsko vrijeme (PV), uredno vrijeme krvarenja (VK) te normalan broj trombocita uz snižene vrijednosti (odnosno aktivnost) faktora VIII i uredne vrijednosti von Willenbrandova faktora. Kod sumnje na hemofiliju A potrebno je diferencijalno dijagnostički isključiti i prisutnost inhibitora faktora VIII što se radi testom miješanja s normalnom plazmom. Kada nakon miješanja uzorka krvi s normalnom plazmom dođe do korekcije aPTV-a, radi se o hemofiliji A, a kada ne dođe, potrebna je daljnja obrada u smislu postojanja eventualnog inhibitora faktora VIII (Slika 6.5.). Danas su moguća i genetska testiranja u vidu dokazivanja vrste mutacije gena za faktor VIII.
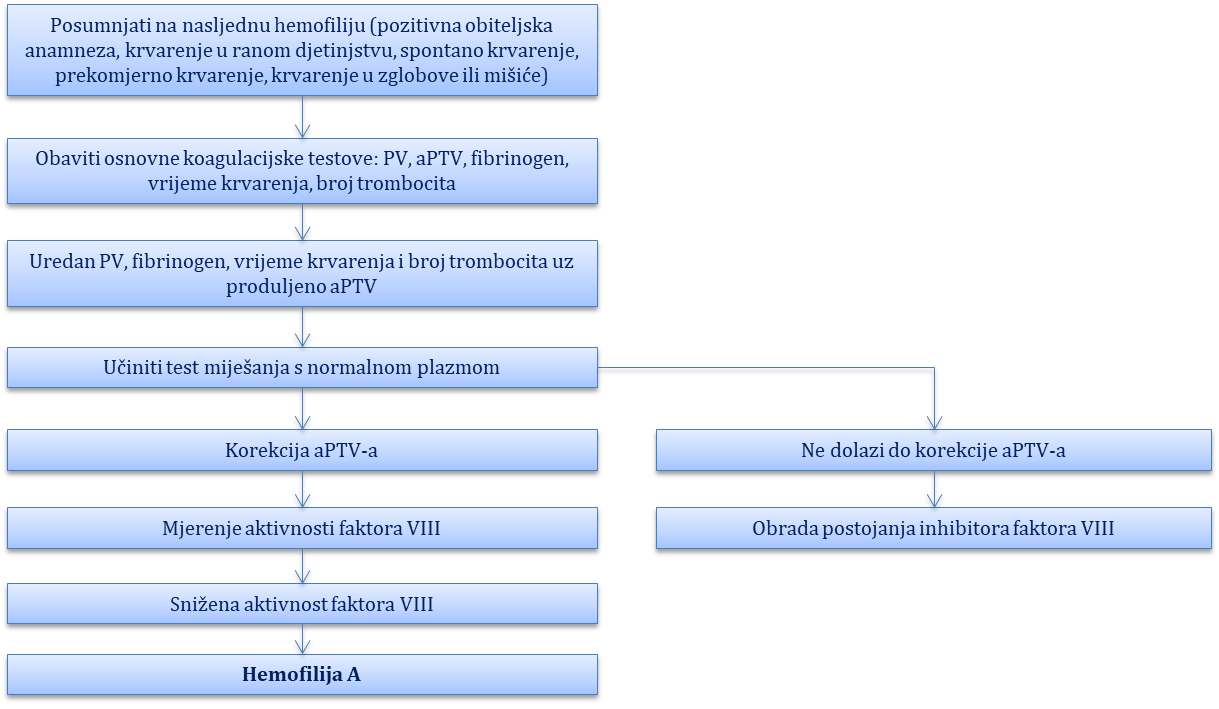
Slika 6.5. Dijagnostički postupak kod sumnje na hemofiliju
Liječenje. Osnovu liječenja bolesnika s hemofilijom A predstavlja epizodna ili profilaktička supstitucijska terapija faktorom VIII. Epizodna primjena podrazumijeva primjenu faktora VIII kod pojave krvarenja, obično unutar dva sata od nastupa krvarenja, dok profilaktička primjena podrazumijeva redovnu supstituciju faktorom VIII (obično tri puta tjedno) kako bi se izbjeglo nastanak krvarenja. Terapijski je pristup individualan i ovisi o težini bolesti (npr. teška hemofilija zahtijeva profilaktičku primjenu), bolesnikovim preferencijama i mogućnostima (faktor VIII se primjenjuje isključivo intravenski, što komplicira njegovu primjenu) i sl.
Današnji standard podrazumijeva primjenu koncentrata faktora VIII, a razlikujemo preparate dobivene iz plazme i preparate dobivene rekombinantnom tehnikom koje se preferira zbog smanjenja rizika od nastanka infekcija i imunizacije. Primjena krioprecipitata i svježe smrznute plazme s ciljem supstitucije faktora VIII danas se ne preporuča. Kod profilaktičkoga liječenja potrebno je osigurati održavanje aktivnosti faktora VIII iznad 1 do 2 %, a to se obično postiže primjenom koncentrata faktora VIII u dozi od 20 do 40 IJ/kg triput tjedno (ovisno o odgovoru bolesnika doze se može povećavati ili snižavati, kao i učestalost primjene). Liječenjem akutnoga krvarenja potrebno je postići urednu vrijednost faktora VIII u krvi (> 50 IU/dL), a treba imati na umu i da je poluvijek života faktora VIII svega 8 do 12 sati, zbog čega se mora davati u više dnevnih doza ili u obliku kontinuirane intravenske infuzije.
Kod blažih oblika hemofilije može biti indicirana i primjena dezmopresina, sintetičkoga analoga vazopresina koji ima sposobnost izazivanja povećanja koncentracije (aktivnosti ) faktora VIII i von Willenbrandova faktora. Može se primjenjivati intranazalno, subkutano ili intravenski, a učinak se vidi kroz nekoliko sati. Može se primjenjivati samo kod bolesnika čije su bazalne vrijednosti faktora VIII veće od 10 IU/dL.
Najznačajnija nuspojava takve terapije razvoj je inhibitora za faktor VIII, a radi se o IgG4 protutijelima koja neutraliziraju funkciju danih faktora VIII. Liječenje krvarenja kod takvih bolesnika otežano je te uključuje primjenu visokih doza faktora VIII, antifibrinolitika (transeksamična kiselina), aktiviranoga rekombinantnog faktora VII (rFVIIa) te aktiviranoga protrombinskog kompleksa (aPCC). Emicizumab je humanizirano monoklonsko modificirano imunoglobulinsko (IgG4) protutijelo bispecifične strukture. Emicizumab premošćuje aktivirani faktor IX i faktor X i tako ponovno uspostavlja funkciju nedostajućega aktiviranog faktora VIII potrebnoga za učinkovitu hemostazu.
Liječenje takvih bolesnika provodi se u odgovarajućim zdravstvenim centrima, a svi bolesnici i članovi obitelji moraju proći odgovarajuću edukaciju. Bolesnici moraju izbjegavati fizičke aktivnosti koje bi mogle rezultirati ozljedom i posljedičnim krvarenjem, a potrebno je izbjegavati i primjenu acetilsalicilne kiseline i nesteroidnih protuupalnih lijekova. Zabranjena je primjena intramuskularnih injekcija kod takvih bolesnika.
Hemofilična artropatija najčešća je i najkarakterističnija komplikacija hemofilije. Ona nastaje kao posljedica krvarenja u zglobnu čahuru i posljedične upale koja dovodi do destrukcije hrskavice, fibroze i konačno ankiloze zgloba. Početni znakovi uključuju otok, bol i crvenilo koje potom zamjenjuju ograničenje pokreta, ukočenost zgloba i kontrakture. Dijagnoza se potvrđuje nekom od slikovnih metoda (rendgenogram, ultrazvuk, magnetna rezonanca). Terapija uključuje profilaktičku primjenu faktora VIII, metode fizikalne terapije, a u najtežim oblicima indicirano je i operativno liječenje.
Prognoza. Uz pridržavanje osnovnih preporuka i susptitucijsko liječenje bolesnici imaju dobru prognozu i normalan očekivani životni vijek te su smrtni ishodi vezani uz krvarenje izuzetno rijetki.
HEMOFILIJA B
Definicija. Hemofilija B označava nasljednu sklonost krvarenju koja se javlja kao posljedica nedostatka ili smanjenje funkcije koagulacijskoga faktora IX. Radi se o glikoproteinu koji nastaje u jetri i čija je sinteza ovisna o vitaminu K, a poluvijek života u plazmi iznosi 18 do 24 sata. Učestalost hemofilije B znatno je rjeđa od hemofilije A, a procjenjuje se da se pojavljuje kod jednoga na 30 000 muške djece.
Etiopatogeneza. Hemofilija B nastaje kao posljedica mutacije gena za faktor IX koji se, kao i gen za faktor VIII, nalazi na dugom kraku X kromosoma (Xq27), a prenosi se recesivnim X-vezanim putem tako da vrijede ista pravila kao i za hemofiliju A. Posljedično manjku ili nefunkcionalnosti faktora IX koji pripada unutarnjem putu zgrušavanja izostaje odgovarajuća aktivacija faktora X čime otpočinje zajednički put zgrušavanja.
Klinička slika i dijagnoza. Kliničke manifestacije hemofilije B identične su onima kod bolesnika s hemofilijom A te se ta dva poremećaja ne može razlikovati na temelju kliničke slike. Novija istraživanja pokazala su da je učestalost krvarenja i hemofilične artropatije veća kod bolesnika s hemofilijom A u odnosu na bolesnike s hemofilijom B s istom razinom bolesti (blaga, umjerena, teška). Teški oblici bolesti obilježeni su razinom aktivnosti faktora IX manjom od 1 %, umjereni oblici između 1 i 5 %, a blagi oblici imaju razinu aktivnosti između 5 i 40 %. Laboratorijski nalazi identični su kao i kod bolesnika s hemofilijom A (produljen aPTV, uredan PV, uredan von Wilenbrandov faktor) samo što je kod ovih bolesnika smanjena razina (aktivnost) faktora IX. Genetska testiranja omogućavaju otkrivanje mutacije gena za faktor IX, kojih je do sada opisano više od nekoliko stotina.
Liječenje podrazumijeva profilaktičku ili epizodnu primjenu koncentrata faktora IX (plazmatski, rekombinantni). Profilaktička primjena faktora IX obično se provodi kroz dvije tjedne aplikacije u dozi od 20 do 40 IJ/kg. Pojava inhibitora faktora IX znatno je rjeđa nego za faktor VIII. Primjena dezmopresina kod ovih bolesnika nije opravdana jer ne dovodi do povećanja serumske koncentracije faktora IX.
Prognoza takvih bolesnika uz pravilno liječenje i pridržavanja preporuka jednaka je kao i kod bolesnika koji boluju od hemofilije A.
VON WILLENBRANDOVA BOLEST
Definicija. Von Willenbrandova bolest predstavlja autosomno dominantnu nasljednu bolest koja se manifestira kao sklonost krvarenju, a nastaje uslijed nedostatka ili smanjene funkcije von Willenbradova faktora (vWF), multimernoga glikoproteina kojega u najvećoj mjeri stvaraju endotelne stanice. vWF ima dvojaku ulogu jer sudjeluje kao posrednik u adheziji i agregaciji trombocita te kao nosač i stabilizator faktora VIII u plazmi. Radi se o najčešćem nasljednom obliku hemoragijske dijateze s procijenjenom incidencijom od 1 na 800 do 1000 osoba u općoj populaciji.
Etiopatogeneza. Von Willenbrandova bolest nastaje uslijed kvantitativnoga ili kvalitativnog poremećaja u sintezi vWF-a, a što je posljedica genetskih promjena gena za vWF koji se nalazi na kromosomu 12. Sinteza vWF-a najvećim se dijelom odvija u endotelnim stanicama (90 %), a tek se manji dio (10 %) sintetizira u megakariocitima koštane srži te on ostaje pohranjen u trombocitima. vWF se luči kao odgovor na ozljedu, stres ili nakon primjene nekih lijekova (npr. dezmopresin). Izlučeni veliki multimeri vWF-a imaju izraženu prokoagulacijsku aktivnost te se u normalnim okolnostima oni cijepaju na manje fragmente uz pomoć ADAMTS13 metaloproteinaza (opisano u poglavlju o TTP-u). Ovisno o mutaciji gena za vWF, može ga se stvarati u manjoj količini (kvantitativni poremećaj) ili stvoreni vWF može biti afunkcionalan (kvalitativni poremećaj). Zbog toga dolazi do poremećaja funkcije trombocita (slabija adhezija za oštećeni vaskularni endotel, smanjenja sposobnost agregacije trombocita), a smanjena je i njegova funkcija kao nosača i stabilizatora faktora VIII, zbog čega se on ubrzano metabolizira i nestaje iz cirkulacije. Sve opisane okolnosti rezultiraju povećanom sklonošću krvarenju.
Klasifikacija. S obzirom na karakteristike, razlikujemo tri oblika von Willenbrandove bolesti. Tip 1 najčešći je oblik koji se susreće kod oko 75 do 80 % bolesnika, a predstavlja kvantitativni poremećaj, uslijed kojega bolesnici imaju smanjene plazmatske koncentracije vWF-a i faktora VIII, a težina koagulopatije korelira s vrijednostima vWF-a i faktora VIII. Tip 2 je rjeđi i javlja se kod oko 20 do 25 % oboljelih, a predstavlja kvalitativni poremećaj vWF-a. Njegova je plazmatska vrijednost uredna, dok je razina faktora VIII snižena, a ovisno o osobinama poremećaja dodatno se razlikuju četiri podoblika. Tip 3 je najteži i najrjeđi oblik (< 1 % oboljelih) karakteriziran potpunim izostankom stvaranja vWF-a i izraženom sklonošću krvarenju, a za razliku od tipa 1 i 2, nasljeđuje se autosomno recesivnim putem.
Klinička slika. Kliničke manifestacije i težina hemoragijske dijateze ovise ponajprije o težini bolesti. Tip 3 von Willenbrandove bolesti obilježen je teškim, učestalim i po život opasnim krvarenjima koja se manifestiraju već u ranom djetinjstvu. Tip 1 i 2 obično su praćeni krvarenjima u kožu i sluznicu koja se ovisno o težini bolesti javljaju spontano ili nakon traume. Također, ovi bolesnici imaju povećani rizik od teških krvarenja povezanih s kirurškim i drugim invazivnim postupcima. Krvarenja u zglobove i hemofilična artropatija nisu karakteristični za ove bolesnike.
Dijagnoza se postavlja na osnovi kliničke slike, anamnestičkih podataka (poremećaj zgrušavanja krvi kod bližih rođaka) te laboratorijskih nalaza. U osnovnim laboratorijskim nalazima broj trombocita je uredan, protrombinsko vrijeme uredno, produljeno je vrijeme zgrušavanja i, ovisno o vrijednostima faktora VIII, produljeno aktivirano parcijalno tromboplastinsko vrijeme. Konačna dijagnoza postavlja se specifičnom laboratorijskom obradom koja uključuje određivanje aktivnosti i antigena vWF, određivanje vrijednosti faktora VIII te ispitivanje agregacije trombocita (RIPA, engl. ristocetin-induced platelet aggregation), kako je navedeno u Tablici 6.45.
|
Tablica 6.45. Dijagnoza von Willenbrandove bolesti
|
|
Tip
|
Aktivnost vWF-a
|
Antigen vWF
|
Faktor VIII
|
RIPA
|
|
Tip 1
|
↓
|
↓
|
↓ ili N
|
↓
|
|
Tip 2A
|
↓↓
|
↓
|
↓
|
↓
|
|
Tip 2B
|
↓↓
|
↓
|
↓
|
↑
|
|
Tip 2N
|
↓
|
↓
|
↓
|
↓
|
|
Tip 2M
|
N
|
N
|
↓↓
|
N
|
|
Tip 3
|
↓↓↓
|
↓↓↓
|
↓↓↓
|
↓↓↓
|
Liječenje bolesnika s von Willenbrandovom bolesti temelji se na profilaksi i/ili liječenju krvarenja. Profilaksa se provodi kod većine bolesnika neposredno prije te nakon invazivnih zahvata i procedura, rijetko je potrebna redovita, odnosno kontinuirana profilaksa. U tom pogledu na raspolaganju stoje tri opcije: 1) primjena dezmopresina koji potiče oslobađanje vWF-a pohranjenoga u endotelnim stanicama; 2) primjena koncentrata faktora VIII i vWF-a dobivenih iz plazme ili rekombinantnom tehnologijom te 3) primjena ostalih hemostatskih sredstava koja potiču zgrušavanje krvi, a ne djeluju izravno na faktor VIII ili vWF (npr. primjena transeksamične kiseline). Pri teškim krvarenjima, u nedostatku koncentrata vWF-a kao njegov se izvor može koristiti i krioprecipitat, ali ga treba ograničiti i čim prije zamijeniti koncentratom vWF-a. Profilaksu kod većih zahvata (torakalne, abdominalne operacije) treba provoditi 7 do 14 dana, dok kod manjih zahvata (biopsije, stomatološki zahvati, centralni kateteri) ona traje od jednog do pet dana. Provodi se koncentratom vWF-a obično s početnom dozom od 40 do 60 U/kg u nastavku s 20 do 40 U/kg svakih 8 do 24 sata.
STEČENI POREMEĆAJI
Najčešći stečeni poremećaji koji mogu rezultirati hemoragijskom dijatezom su deficijencija vitamina K, jetrena insuficijencija, diseminirana intravaskularna koagulopatija te stečena hemofilija.
DEFICIJENCIJA VITAMINA K
Vitamin K jedan je od ključnih vitamina koji sudjeluje u sintezi faktora II, VII, IX i X te proteina C i S, a sudjeluje u gama-karboksilaciji aminokiseline glutamina, što je nužno za vezanje kalcija i njihovu aktivaciju. Posljedično nedostatku vitamina K, navedeni faktori ne posjeduju karboksilnu skupinu, ne mogu vezati kalcij te se ne mogu aktivirati, što dovodi do manjkavosti koagulacijske kaskade i posljedične sklonosti krvarenju. Dva su osnovna izvora vitamina K u organizmu, a to su enteralni unos putem hrane te sinteza unutar probavnoga sustava uslijed djelovanja bakterijske flore crijeva, s time da je za apsorpciju iz crijeva potrebna prisutnost žučnih soli. Prema tome, četiri su osnovna moguća uzroka manjka vitamina K: 1) smanjen enteralni unos vitamina K putem hrane, 2) malapsorpcija vitamina K kao posljedica različitih oboljenja probavnoga sustava, 3) smanjena endogena produkcija zbog poremećaja crijevne flore što se viđa kod bolesnika na antibiotskoj terapiji ili uslijed primjene kemoterapeutika te 4) peroralna primjena antagonista vitamina K (varfarin) koji se koriste u antikoagulantnom liječenju. U laboratorijskim nalazima ovih bolesnika obično se viđa produljeno protrombinsko vrijeme (PV, INR), a pri težim oblicima produljeno može biti i aktivirano parcijalno tromboplastinsko vrijeme (aPTV). Liječenje se sastoji od primjene vitamina K, bilo peroralno bilo intravenski (supkutana i intramuskularna primjena se ne preporučuju). Peroralna primjena je rijetka, primjenjuje se 5 do 10 mg vitamina K (fitonadion) na dan, a normalizacija PV-a očekuje se kroz 18 do 24 sata. Intravenska primjena je češća, osobito kod bolesnika s izraženom hemoragijskom dijatezom i produljenim PV-om, kao i kod bolesnika s malapsorpcijom. Tim se načinom primjenjuju manje doze (1 - 5 mg/dan), a ispravak PV-a očekuje se znatno brže (kroz nekoliko sati). U slučaju jakoga krvarenja uz intravenski vitamin K potrebna je i primjena svježe smrznute plazme.
JETRENA INSUFICIJENCIJA
Poremećaji zgrušavanja krvi, tj. sklonost krvarenju, često prate bolesnike s narušenom funkcijom jetre, a s njome povezano krvarenje odgovorno je za oko 20 % uzroka smrti bolesnika s uznapredovalom bolešću jetre. Koagulopatija se kod takvih bolesnika razvija iz više razloga od kojih je najznačajnija smanjena sinteza koagulacijskih faktora uslijed destrukcije hepatocita, a tome pridonose i drugi čimbenici, kao što su smanjena apsorpcija vitamina K zbog intrahepatične ili ekstrahepatične kolestaze (za apsorpciju vitamina K nužna je prisutnost žuči u crijevu), trombocitopenije uslijed splenomegalije kao posljedice portalne hipertenzije koju nalazimo u ovih bolesnika te funkcionalnih poremećaja trombocita i fibrinogena uslijed toksemije. Osim krvarenja u kožu i sluznice, kod takvih su bolesnika česta i krvarenja u probavni sustav zbog portalne gastropatije i varikoziteta jednjaka. Iako kod takvih bolesnika nalazimo patološke vrijednosti i PV-a, aPTV-a i fibrinogena, najosjetljivijim se smatra PV, koji se koristi i u prognostičke svrhe. Primjena vitamina K može imati povoljan učinak tek kod bolesnika s blažim oblicima bolesti, dok je u uznapredovaloj bolesti jetre njegova primjena beskorisna. Temelj liječenja koagulopatije kod tih bolesnika čini primjena svježe smrznute plazme i trombocita kao privremeno rješenje dok je dugotrajnije rješenje transplantacija jetre.
DISEMINIRANA INTRAVASKULARNA KOAGULOPATIJA
Diseminirana intravaskularna koagulopatija opisana je u poglavlju „Anemija, trombocitopenija i koagulopatija u kritično oboljelih bolesnika“.
STEČENA HEMOFILIJA
Stečena hemofilija označava rijetko stanje karakterizirano stvaranjem i prisutnošću autoantitijela na faktore zgrušavanja, najčešće na faktor VIII. Najčešće se susreće kod starijih osoba, i to onih koji u podlozi imaju malignu ili autoimunu bolest. Klinička slika je varijabilna i ovisi o stupnju inhibicije faktora koagulacije. Kako se najčešće radi o autoantitijelima na faktor VIII, u koagulogramu nalazimo tipičan obrazac hemofilije A: produljeno aPTV uz uredan PV, trombocite i fibrinogen. Test miješanja s plazmom (opisano gore u tekstu) omogućava razlikovanje stvarnoga manjka faktora VIII od prisutnosti inhibitora. Definitivna dijagnoza postavlja se utvrđivanjem postojanja inhibitora faktora VIII. Osnovu liječenja čine kortikosteroidi ili rituksimab kod najtežih oblika, a kod pojave krvarenja koristi se koncentrat aktiviranoga protrombinskog kompleksa ili rekombinantni aktivator faktora VII.
SKLONOST ZGRUŠAVANJU KRVI ILI TROMBOFILIJA
Definicija. Trombofilija je naziv za nasljedni ili stečeni poremećaj sustava zgrušavanja krvi koji izaziva sklonost zgrušavanju krvi, odnosno nastanku tromboze. Bolesnici koji imaju neku od trombofilija ubrajaju se u rizičnu skupinu za obolijevanje od tromboembolijskih incidenata (duboka venska tromboza, plućna embolija) koji čine glavnu kliničku manifestaciju, ali imaju i druge rizike, kao što su rekurentni spontani pobačaji kod žena, nekroza kože izazvana varfarinom i sl. Tromboze se kod takvih bolesnika često pojavljuju na neuobičajenim mjestima (npr. duboka venska tromboza gornjih ekstremiteta) te u mlađoj životnoj dobi (prije 45. godine života). U nastavku teksta bit će opisane najčešće nasljedne i stečene trombofilije te klinički pristup bolesnicima s trombofilijama.
Najznačajnije trombofilije u kliničkoj praksi su deficit antitrombina III, deficit proteina C, deficit proteina S, rezistencija na aktivirani protein C, mutacija gena za protrombin G20210A, hiperhomocisteinemija, antifosfolipidni sindrom te mutacija inhibitora aktivatora plazminogena.
Deficit antitrombina III. Antitrombin III predstavlja glavni fiziološki inhibitor trombina, ali i drugih aktiviranih koagulacijskih faktora, kao što su faktori IX, X, XI i XII. Gen za antitrombin III nalazi se na kromosomu 1, a sinteza se odvija u stanicama vaskularnoga endotela. Učinak antitrombina III ostvaruje se kroz ireverzibilno vezanje na serinske proteaze aktiviranih koagulacijskih faktora, a nastali kompleks dovodi do njihove inaktivacije (djelovanjem heparina to se vezivanje ubrzava i pojačava, na čemu se i temelji terapijski antikoagulantni učinak heparina). Homozigotni manjak antitrombina III rezultira apsolutnim manjkom antitrombina III i nespojiv je sa životom, dok se kod heterozigota bilježi veći rizik od nastanka tromboebolijskih incidenata. Do sada je dokazano više genetskih defekata koji mogu dovesti do smanjenoga stvaranja antitrombina III i/ili stvaranja disfunkcionalnoga antitrombina III, a prenose se autosomno dominantno. Razlikujemo dva oblika bolesti: tip I i tip II. Deficit antitrombina tip I označava stanje u kojemu nalazimo smanjenu količinu antitrombina III uz njegovu urednu funkciju, dok u tipu II nalazimo urednu količinu antitrombina III, ali je njegova funkcija poremećena. Funkcija antitrombina III izražava se mjerenjem njegove aktivnosti, dok se njegova količina utvrđuje određivanjem količine antigena antitrombina III u plazmi. Učestalost heterozigotnih mutacija gena za antitrombin III u općoj se populaciji kreće između 1:2000 i 1:5000, a većina bolesnika cijeloga života ostaje asimptomatska. Dijagnoza se potvrđuje određivanjem funkcionalnosti (aktivnosti) antitrombina III te određivanjem koncentracije njegova antigena u plazmi, a na temelju tih vrijednosti razlikujemo gore opisana dva tipa bolesti: bolesnici s tipom I imaju smanjene vrijednosti antigena, kao i aktivnosti antitrombina III, dok bolesnici s tipom III imaju uredne vrijednosti antigena uz smanjenu aktivnost antitrombina III. Stečene bolesti i stanja koja se povezuju s manjkom antitrombina III su bolesti jetre, nedavna ili aktivna tromboza, diseminirana intravaskularna koagulopatija, liječenje heparinom, nefrotski sindrom te liječenje estrogenima.
Deficit proteina C. Protein C je snažan koagulacijski inhibitor koji inaktivira prethodno aktivirane faktore V i VIII. Gen za protein C nalazi se na drugom kromosomu, a sama sinteza proteina C u najvećoj se mjeri odvija u jetri, a u nešto manjoj mjeri i u Leydingovim stanicama testisa i u epididimisu. Da bi protein C ostvario svoju ulogu i inaktivirao faktore V i VIII, on se najprije mora aktivirati, što se zbiva na površini endotelnih stanica uz posredovanje kalcija, trombina i trombomodulina. U nedostatku proteina C dolazi do pojačane aktivnosti faktora V i VIII što u konačnici rezultira povećanom sklonošću nastanku tromboze. Do danas je opisano više stotina vrsta mutacija gena za protein C, a nasljeđuju se autosomno dominantnim putem. Homozigotni manjak proteina C je, kao i manjak antitrombina III, nespojiv sa životom, dok se učestalost heterozigota u općoj populaciji kreće između 1:200 i 1:500. Kao i oboljeli od drugih nasljednih trombofilija, i ovi bolesnici imaju povećan rizik od nastanka tromboembolijskih incidenata te se deficit proteina C može otkriti kod oko 5 % bolesnika koji se prezentiraju recidivnim trombozama. Prema ekspresiji bolesti i laboratorijskim nalazima razlikuju se dva tipa bolesti: tip I i tip II. Kod bolesnika s tipom I, koji je znatno češći, nalazimo sniženu aktivnost, kao i smanjenu ukupnu količinu proteina C, kao posljedicu smanjene sinteze proteina C, dok je kod bolesnika s tipom II aktivnost smanjena uz urednu količinu proteina C, što nastaje kao rezultat očuvane sinteze disfunkcionalnoga proteina C. Manjak proteina C može se naći i kod nekih stečenih bolesti i stanja, kao što su bolesti jetre, primjena oralnih antikoagulanasa, manjak vitamina K, nedavna ili aktivna tromboza, diseminirana intravaskularna koaguopatija, teška infekcija i sl.
Deficit proteina S. Protein S djeluje kao kofaktor aktiviranoga proteina C u inhibiciji koagulacijske kaskade djelujući na aktivirane faktore V i VIII. Protein S u plazmi cirkulira vezan na proteinu koji veže C4b, a samo slobodni dio ujedno je i aktivan, tako da na samu aktivnost proteina S utječe i vrijednost navedenoga proteina u plazmi. Nasljedni manjak proteina S nalazimo u oko 0,7 % populacije. Homozigoti za manjak toga proteina klinički se manifestiraju ubrzo po rođenju, dok heterozigoti imaju slične kliničke manifestacije kakve nalazimo i kod drugih trombofilija. Kao i kod prethodno opisanih trombofilija, razlikujemo dva tipa bolesti: tip I (smanjena količina i funkcija proteina S) i tip II (smanjena funkcija proteina S uz uredne količinske vrijednosti). Stečeni manjak proteina S može se naći u bolestima i stanjima jednakima kao i kod proteina C.
Rezistencija na aktivirani protein C. Danas se smatra da je rezistencija faktora V i VII na aktivirani protein C jedna od najčešćih nasljednih trombofilija koje se susreću u kliničkoj praksi te se ona može naći kod oko 20 do 40 % bolesnika s recidivirajućim tromboembolijskim incidentima. Taj poremećaj nastaje kao posljedica genetskih poremećaja koji onemogućavaju aktiviranom proteinu C da inaktivira aktivirane faktore V i VII. U najvećem broju slučajeva (> 95 %) radi se o mutaciji faktora V Leiden (G1691A), dok se rjeđe radi o mutaciji faktora V Cambrige ili mutaciji faktora V Hong Kong. Rezistencija na aktivirani protein C određuje se tako da se odredi protrombinsko vrijeme u klasičnom uzorku krvi, a potom u uzroku krvi u koji je dodan aktiviran protein C jer dodatak aktiviranoga proteina C u normalnim okolnostima rezultira produljenjem protrombinskog vremena. Sama dijagnoza potvrđuje se dokazivanjem specifične mutacije PCR tehnikom.
Mutacija gena za faktor V Leiden najčešći je nasljedni poremećaj koagulacije u populaciji bijele rase, a ime je dobila po gradu Leidenu u Nizozemskoj gdje je prvi put identificirana 1994. godine. Genetski je izazvana točkastom mutacijom na eksonu 10 F5 gena koja mijenja šifru za aminokiselinu arginin (normalan gen) u glutamin (mutiran gen). Kod ljudi bez te mutacije faktor V igra važnu ulogu u aktivaciji trombina koji opet ima važnu ulogu kod formiranja fibrina iz fibrinogena, koji čini većinu ugruška. Defektan faktor V diktiran od strane mutiranoga gena ne može biti deaktiviran proteinom C što dovodi do posljedične hiperkoagulacije. Važno je napomenuti da je to dominantna mutacija, tako da ljudi sa samo jednom kopijom defektnoga gena mogu imati klinička očitovanja. Oko 30 % pacijenata s dubokom venskom trombozom i/ili plućnom embolijom pokazuje tu mutaciju. Žene s tom mutacijom imaju povišen rizik od spontanoga pobačaja ili od rođenja mrtvorođenčeta zbog stvaranja ugrušaka u posteljici ili pupčanoj vrpci, kao i u samome fetusu ako je dijete naslijedilo gen.
Mutacija gena za protrombin G20210A. Opisana mutacija gena za protrombin rezultira povećanom razinom protrombina u krvi što ujedno povećava rizik od nastanka tromboze. Tu se mutaciju može naći kod oko 1 % opće populacije i oko 10 % osoba koje razviju tromboembolijski incident. Dijagnoza se potvrđuje genetskim testiranjem (PCR).
Hiperhomocisteninemija. Povišene vrijednosti homocisteina povezane su s povećanom sklonošću prema razvoju arterijskih i venskih tromboza. Može biti nasljedna ili stečena. Nasljedni oblik homocisteinemije najčešće je povezan s MTHFR mutacijom, a prisutna je kod 5 do 15 % pripadnika opće populacije. Homozigoti s tom mutacijom imaju povišene vrijednosti homocisteina i sklonost razvoju tromboza, dok su heterozigoti asimptomatski. Stečenu hiperhomocisteinemiju možemo naći kod zatajenja bubrega, hipotireoze, malignih oboljenja, manjka vitamina B skupine te vezano uz primjenu određenih lijekova (kortikosteroidi, fenitoin, ciklosporin, metotreksat). U kliničkoj se praksi po otkrivanju povišenih vrijednosti homocisteina u krvi najprije određuje razina vitamina B12 i folne kiseline koji su najčešći uzrok toga, a ako su njihove razine uredne, sumnja se na genetski defekt - točkastu mutaciju C677T gena za enzim MTHFR.
Antifosfolipidni sindrom dijagnosticira se dokazivanjem lupusnoga antikoagulansa (LAC). LAC inhibira fosfolipide u reagensu za određivanje aPTV-a, zbog čega je on produžen (paradoksalno, jer produljen aPTV je povezan s krvarenjem, a ne s trombozama). Klinička slika, produljeni aPTV i pozitivan LAC osiguravaju dijagnozu antifisfolipidnoga sindroma.
Mutacija inhibitora aktivatora plaziminogena (PAI-1 4G/4G) povezana je s tri do pet puta većim rizikom od razvoja duboke venske tromboze u odnosu na osobe koje imaju alele 5G/5G ili 4G/5G.
Kliničke manifestacije. Najčešće kliničke manifestacije nasljednih trombofilija su duboka venska tromboza donjih ekstremiteta i plućna tromboembolija. Osim toga, takvi bolesnici imaju sklonost nastanku tromboza neuobičajenih lokalizacija (npr. gornji ekstremiteti), kao i tromboza splanhičkih i drugih krvnih žila (tromboza vene porte, tromboza mezenterijalnih vena ili arterija, koronarna ili cerebralna tromboza). Trombotski incidenti kod takvih bolesnika pokazuju sklonost recidiviranju, pojavnost u mlađoj životnoj dobi te pojavnost bez posebnoga provocirajućeg događaja (tzv. neprovocirajuća tromboza). Bolesnici s deficitom proteina C i S pokazuju i značajnu sklonost razvoju varfarinom inducirane nekroze kože, s obzirom na to da je za sintezu proteina C i S nužan vitamin K. Žene s nasljednim trombofilijama imaju povećan rizik od pojave recidivirajućih spontanih pobačaja. Prema učestalosti nastanka trombotskih incidenata, trombofilije dijelimo na visokorizične i niskorizične, kako je to prikazano u Tablici 6.46.
|
Tablica 6.46. Klasifikacija nasljednih trombofilija
|
|
Visokorizične trombofilije
|
Niskorizične trombofilije
|
|
Nedostatak antitrombina
Homozigot za mutaciju faktora V Leiden
Istodobna prisutnost heterozigota za mutaciju protrombina i faktora V Leiden
Homozigot za mutaciju protrombina
|
Heterozigot za mutaciju faktora V Leiden
Heterozigot za mutaciju protrombina
Nedostatak proteina C
Nedostatak proteina S
|
Dijagnostički pristup i obrada. Probir bolesnika na trombofilije nije rutinski indiciran, nego se provodi samo u određenim okolnostima kao što su: 1) pojava tromboze u mlađoj životnoj dobi (< 40 godina), 2) pojava tromboze kod bolesnika s pozitivnom obiteljskom anamnezom na trombotske događaje, 3) recidivirajuće tromboze koje se ne mogu povezati s jasnim uzrokom, 4) tromboze neuobičajenih lokalizacija (visceralne vene, kavernozni sinusi), 5) udružena pojava arterijskih i venskih tromboza, 6) učestali spontani pobačaji kod žena, 7) pojava tromboze nakon trivijalnih događaja (minimalna trauma), 8) bolesnici s učestalim površinskim tromboflebitisima u odsutnosti varikoznoga sindroma te 9) pojava nekroze kože nakon uvođenja varfarina u terapiju. Probir bolesnika na trombofilije ima smisla samo ako su prethodno isključenja maligna oboljenja, nedavni veliki kirurški zahvati, trudnoća i porođaj te dugotrajno mirovanje i imobilizacija.
Probir na trombofilije uključuje, osim klasičnoga koagulograma, sljedeće pretrage: 1) određivanje rezistencije na protein C, 2) određivanje vrijednosti proteina C, 3) određivanje vrijednosti proteina S, 4) određivanje vrijednosti antitrombina III, 5) određivanje mutacije protrombinskoga gena (G20210A), 6) određivanje mutacije gena faktora V Leiden 7) određivanje vrijednosti homocisteina, 8) određivanje lupusnoga antikoagulansa te 9) mutacije inhibitora aktivatora plaziminogena (PAI-1). Provođenje tih pretraga preporuča se nakon prestanka antikoagulacijskoga liječenja, i to u vremenskom periodu od najmanje dva tjedna s obzirom na to da ovi lijekovi mogu utjecati na vrijednosti većine zatraženih pretraga (ne utječu na nalaze ispitivanja genskih mutacija).
Terapijski pristup. Početno liječenje bolesnika koji razviju trombotski incident, a imaju nasljednu trombofiliju, jednako je kao i kod drugih bolesnika (obično je ona i nepoznata, odnosno nedijagnosticirana), a uključuje razmatranje o eventualnom trombolitičkom liječenju (ovisno o indikaciji), liječenje niskomolekularnim ili klasičnim heparinom uz naknadno peroralno antikoagulacijsko liječenje kroz period od najmanje tri do šest mjeseci. Produljeno antikoagulantno liječenje bolesnika ovisi o više čimbenika među kojima je i rizik od krvarenja (detaljno opisano u poglavlju Duboka venska tromboza), ali kod bolesnika s dva ili više trombotskih incidenata ono bi trebalo biti doživotno. Asimptomatske osobe s poznatom trombofilijom koje nisu imale trombotske incidente ne zahtijevaju profilaktičku antikoagulantnu terapiju, osim u rizičnim situacijama (perioperativno, peripartalno). Zbog opasnosti od razvoja nekroze kože, treba biti na oprezu pri primjeni varfarina kod bolesnika s dokazanim deficitom proteina C ili S (kod primjećivanja promjena na koži odmah je indicirana primjena svježe smrznute plazme ili koncentrata proteina C). Kod bolesnika s deficitom antitrombina III učinak heparina često je oslabljen zbog čega se indicira primjena antritrombina III u obliku koncentrata ili putem svježe smrznute plazme (u nedostatku antitrombina III heparin ne može postići svoj terapijski učinak).
SPLENOMEGALIJA, HIPERSPLENIZAM I OSTALE BOLESTI SLEZENE
S obzirom na to da je slezena najveći limfatični organ u ljudskom tijelu i da su njezini poremećaji često povezani s hematološkim oboljenjima, njezini se patološki poremećaji najčešće opisuju i obrađuju u sklopu hematologije. Najčešći su poremećaji slezene splenomegalija i posljedični hipersplenizam, dok se rjeđe mogu susresti i hiposplenizam, ciste i kongenitalne anomalije slezene.
SPLENOMEGALIJA I HIPERSPLENIZAM
Definicija. Splenomegalija je naziv za povećanje mase slezene koje može biti udruženo i s njezinim pojačanim radom, a tada govorimo o hipersplenizmu. Hipersplenizam je u prvome redu obilježen pojačanim uklanjanjem krvnih stanica iz krvotoka s posljedičnom citopenijom i njezinim posljedicama (anemija, neutropenija i trombocitopenija) te kompenzatornom hiperplazijom koštane srži, a sve se to normalizira nakon splenektomije.
Etiologija. Splenomegalija i hipersplenizam mogu nastati uslijed različitih bolesti i stanja: 1) bakterijske infekcije (endokarditis, bruceloza, sifilis, trbušni tifus, gnojni apsces, tuberkuloza); 2) virusne infekcije (Epstein-Barrov virus, citomegalovirus, virusi hepatitisa); 3) gljivične i parazitarne infekcije (histoplazmoza, toksoplazmoza, malarija, lišmenijaza); 4) benigni poremećaji imunološkoga sustava (reumatoidni artritis, sistemski eritematozni lupus, reakcije preosjetljivosti na lijekove, serumska bolest); 5) maligni poremećaji imunološkoga sustava (leukemije, limfomi, maligna histiocitoza); 6) ostale maligne bolesti (sekundarizmi melanoma, sarkoma, karcinoma); 7) nemaligna hematološka oboljenja (autoimuna hemolitička anemija, hereditarna sferocitoza, talasemija, ekstramedularna hematopoeza); portalna hipertenzija i kongestija (desnostrano srčano popuštanje, bolesti jetre, tromboza jetrenih ili portalne vene); 8) ostali poremećaji (bolesti odlaganja i nakupljanja, infiltrativne bolesti).
Klinička slika. Splenomegalija je kod većine bolesnika asimptomatska pojava te se otkrije tijekom pregleda bolesnika, odnosno palpacije abdomena. Najčešći simptomi povezani sa splenomegalijom su tup bol i osjećaj punine u gornjem lijevom kvadrantu abdomena, osjećaj rane sitosti, a nekada se zbog nadražaja freničkoga živca bolesnici mogu žaliti na bol u lijevom ramenu. Ukoliko bolesnici razviju hipersplenizam, mogu imati i simptome i znakove odgovarajuće citopenije (anemični sindrom, sklonost infekcijama ili sklonost krvarenju). Među najčešćim komplikacijama splenomegalije je infarkt slezene koji obilježava pojava oštroga bola u području gornjega lijevog kvadranta abdomena, što je često praćeno nadražajem frenikusa i ošita, povišenom temperaturom te rupturom slezene koju prati jako intraabdominalno krvarenje i brz razvoj hemoragijskoga šoka.
Dijagnoza splenomegalije najčešće se postavlja već na temelju samoga kliničkog pregleda abdomena, a potvrđuje se slikovnim metodama kao što su ultrazvuk ili kompjuterizirana tomografija abdomena (kompjuterizirana tomografija ima prednost pred ultrazvukom u otkrivanju morfoloških i strukturnih poremećaja unutar same slezene, kao što su apscesi ili sekundarizmi). Kod bolesnika s hipersplenizmom nalazimo različito izražene otklone u krvnoj slici (anemija, trombocitopenija, neutropenija). Glavni dijagnostički izazov predstavlja otkrivanje uzroka splenomegalije, a on se ponajprije zasniva na anamnestičkim podacima, fizikalnom pregledu i osnovnim laboratorijskim nalazima koji upućuju na moguću etiologiju (infektivno oboljenje, hematološko oboljenje, kongestija i sl.). Biopsija slezene se zbog velikoga rizika od komplikacija izuzetno rijetko provodi te se u nejasnim slučajevima, a osobito ako je u podlozi i hipersplenizam, preporuča splenektomija.
Liječenje splenomegalije i hipersplenizma je simptomatsko. Splenektomija (obično laparoskopska) indicirana je kod bolesnika s izraženim manifestacijama hipersplenizma, kao i kod bolesnika s prijetećim ili nastalim komplikacijama kao što su infarkt ili ruptura slezene. Treba imati na umu da su bolesnici nakon splenektomije ugroženi od bakterijskih infekcija uzrokovanih inkapsuliranim sojevima (S. pnumoniae, H. influenzae, N. meningitidis), zbog čega se redovno trebaju cijepiti.
OSTALE BOLESTI SLEZENE
Hiposplenizam je naziv za smanjenju funkciju slezene. Obično je prisutna kod bolesnika nakon splenektomije, a može se naći u sklopu razvojnih poremećaja slezene (ageneza, hipogeneza) te infiltrativnih i drugih bolesti koje zahvaćaju slezenu (amiloidoza, sarkoidoza, tuberkuloza, tumor, višestruki infarkti). Glavne kliničke posljedice hiposplenizma su sklonost infekcijama (posebice uzrokovanim inkapsuliranim bakterijskim sojevima), kao i povećanje broja krvnih stanica (policitoza) zbog izostanka uloge slezene u njihovoj eliminaciji (policitemija uslijed prestanka funkcije slezene obično je prolazne prirode). Temeljni laboratorijski nalaz koji upućuje na hiposplenizam nalaz je Howell Jollyjevih bazofilnih inkluzija u eritrocitima. Liječenje takvih bolesnika je nespecifično i uključuje prevenciju infekcija (cijepljenje) te tromboprofilaksu u slučaju perzistiranja policitemije. Postupci afereze rijetko su potrebni.
Ciste slezene predstavljaju šupljine unutar tkiva slezene koje su obložene epitelom i ispunjene tekućinom, a njihova se incidencija kreće oko 1 %. Razlikujemo prave ciste i pseudociste. Prave ciste prema etiologiji mogu biti parazitarne i neparazitarne (kongenitalne i neoplastične), a karakterizira ih stijenka koju čine epitelne stanice, dok lažne ciste ili pseudociste nemaju epitelnu stijenku, nego samo vezivnu opnu, a one najčešće nastaju nakon trauma, poštednih operacija slezene. Klinički su one najčešće asimptomatske, rijetko mogu uzrokovati bolove ili osjećaj nelagode u gornjem lijevom kvadrantu abdomena. Dijagnoza se postavlja na temelju slikovnih dijagnostičkih metoda, a u diferencijalno dijagnostičke svrhe, osobito kod nejasnih radioloških nalaza, preporuča se serološka obrada za najčešće parazite te tumorski markeri. Većina cista ne zahtijeva liječenje nego ih se prati, a kirurško liječenje može biti opravdano kod bolesnika sa simptomatskom bolesti ili nejasnom etiologijom (npr. sumnja na tumorsku etiologiju). Parazitarne ciste treba kirurški odstraniti.
Razvojne anomalije slezene. Glavne razvojne anomalije slezene su akcesorna i putujuća slezena. Akcesorna slezena označava prekobrojnu slezenu različite veličine, a najčešće je smještena uz hilus slezene ili rep pankreasa (rjeđe u drugim dijelovima trbušne šupljine). Nema veće kliničko značenje, ali nekada može biti uzrok hipersplenizma ili rupture. Scintigrafija daje sigurnu lokalizaciju akcesorne slezene. Putujuća je slezena rijetka anomalija koju karakterizira duga petlja koja izlazi iz hilusa i omogućava mobilnost slezene (nekada i sve do u malu zdjelicu). Nekada može doći do torzije peteljke što može dovesti do kompresije vaskularnih struktura i infarkta slezene, a prezentira se slikom akutnoga abdomena. Od drugih se anomalija spominju ageneza i hipoplazija slezene.
TRANSPLANTACIJA KRVOTVORNIH MATIČNIH STANICA
Definicija. Transplantacija krvotvornih matičnih stanica predstavlja terapijski postupak kojime se manje vrijedne (bolesne) krvotvorne matične stanice zamjenjuju zdravim matičnim stanicama dobivenima iz koštane srži, periferne ili umbilikalne krvi u bolesnika kod kojega je prethodno provedeno mijeloablativno liječenje (kemoterapija, radioterapija).
Osnovni princip takvoga liječenja podrazumijeva najprije uništavanje bolesnoga (malignog) klona intenzivnim kemoterapijskim i radioterapijskim tretmanom, nakon čega se bolesniku reinfundiraju zdrave krvotvorne matične stanice iz kojih će započeti normalna zdrava mijelopoeza. Osim u liječenju hematoloških tumora, taj se način liječenja sve češće primjenjuje i u liječenju drugih hematoloških i imunoloških poremećaja.
Cijeli se proces transplantacije krvotvornih matičnih stanica može podijeliti u tri faze: 1) predtransplantacijska faza, 2) postupak transplantacije te 3) posttransplantacijska faza.
Oblici transplantacije krvotvornih matičnih stanica. S obzirom na izvor, odnosno porijeklo zdravih krvotvornih matičnih stanica, razlikujemo tri oblika transplantacije: 1) alogena transplantacija (davatelj i primatelj su različite HLA-podudarne osobe), 2) singena transplantacija (davatelj i primatelj su jednojajčani blizanci) te 3) autologna transplantacija (davatelj je sam bolesnik).
Prema tome, izvor krvotvornih matičnih stanica mogu biti: alogenična koštana srž ili periferna krv HLA-podudarnoga srodnika, alogenična koštana srž ili periferna krv nesrodnoga HLA-podudarnog darivatelja, singenična koštana srž ili periferna krv (od jednojajčanoga blizanca), autologna koštana srž ili periferna krv te krv iz pupkovine.
S obzirom na to da su kod singene i autologne transplantacije matičnih stanica davatelj i primatelj potpuno HLA-podudarni, kažemo da se radi o imunološki inertnoj transplantaciji, dok je alogena transplantacija matičnih stanica praćena rizikom od imunološki posredovanih reakcija, kao što je reakcija odbacivanja transplantata ili reakcija transplantata protiv domaćina.
Prijetransplantacijska faza obuhvaća sve radnje koje se odvijaju prije samoga postupka transplantacije, a uključuju postavljanje same indikacije za transplantaciju, izbor darivatelja, odnosno izvora krvotvornih matičnih stanica te pripremu bolesnika za samu transplantaciju.
Indikacije za transplantaciju krvotvornih matičnih stanica ne predstavljaju samo maligne bolesti hematopetskoga sustava nego se ona koristi i kod drugih hematoloških oboljenja (aplastična anemija, paroksizmalna noćna hemoglobinurija, Fanconijeva anemija, izolirana aplazija crvene loze), solidnih malignih oboljenja (neuroblastom, rabdomiosarkom) te kod imunoloških i drugih poremećaja (mukopolisaharidoze, primarne imunodeficijencije). Pri postavljanju indikacije za takav način liječenja treba uzeti u obzir i dob i kondicijsko stanje bolesnika (Karnofskyjev skor, ECOG status), s obzirom na to da se radi o vrlo agresivnome načinu liječenja koje zahtijeva dobro kondicijsko stanje bolesnika.
Izbor darivatelja odnosno izvora krvotvornih matičnih stanica ovisi o više čimbenika. Kod autologne transplantacije matičnih stanica njihovo se prikupljanje vrši iz periferne krvi samoga bolesnika nakon njihove mobilizacije iz koštane srži, što se provodi primjenom rekombinantnih čimbenika rasta (čimbenik stimulacije granulocitnih kolonija ili čimbenik stimulacije granulocitno-makrofagnih kolonija). Nakon mobilizacije se matične stanice skupljaju postupkom afereze te se separiraju, pročišćavaju i zamrzavaju do reinfundiranja u bolesnika, što se vrši nakon provedene mijeloablacije. Kod alogene transplantacije matičnih stanica one se uzimaju od HLA-podudarnoga srodnika ili nesrodnika, i to postupkom punkcije koštane srži nakon čega se koštana srž darivatelja stavlja u poseban medij, centrifugira i filtrira, ili postupkom afereze matičnih stanica iz periferne krvi nakon prethodne mobilizacije iz koštane srži. U svrhu utvrđivanja HLA-podudarnosti moraju se izvršiti sljedeći testovi: 1) tipizacija HLA-alela primatelja i davatelja, 2) određivanje broja i vrste HLA-protutijela u serumu primatelja te 3) neposredno pred transplantaciju test križne reakcije između primatelja i davatelja. Kao i kod alogene transplantacije, izvor matičnih stanica kod singene transplantacije može biti koštana srž ili periferna krv, ali se prije toga ne moraju provoditi HLA-tipizacija i drugi imunološki testovi.
Mijeloablativno liječenje dio je predtranslantacijske obrade bolesnika. Primjenom kemoterapije i radioterapije nastoji se u što većoj mjeri smanjiti broj tumorskih stanica (a time se smanjuje i broj zdravih stanica), kao i postići supresiju bolesnikova imunološkoga sustava čime se smanjuje rizik od odbacivanja presatka. Osim toga načina kondicioniranja bolesnika, danas postoji i tzv. kondicioniranje smanjenoga intenziteta (RCI, engl. reduced intensity conditioning), pri čemu se nastoji postići jaka i dugotrajna imunosupresija primatelja primjenom nižih doza zračenja i kemoterapeutika s ili bez primjene antitimocitnoga globulina, s ciljem omogućavanja prihvaćanja stanica iz presatka, a tek potom eliminacija ostatnh tumorskih stanica imunološkim, a ne izravnim citotoksičnim učinkom, za što su odgovorni T-limfociti koji posreduju reakciju presatka protiv tumora (GvT, engl. graft versus tumor effect). Zbog postizanja potpunoga efekta tim se bolesnicima povremeno dodatno u određenim vremenskim intervalima infundiraju darivateljevi limfociti (DLI, engl. donor lymphocytes infusion).
Transplantacija se odvija izravnim intravenskim infundiranjem pripremljenih matičnih stanica u bolesnika što se provodi kroz nekoliko sati putem središnjega venskog katetera.
Poslijetrasplantacijska faza podrazumijeva intenzivan nadzor i praćenje bolesnika s obzirom na to da se oni početno nalaze u fazi pancitopenije i potencijalnih nuspojava koje ona nosi. U tom je ranom periodu bitno provoditi potporno liječenju u obliku transfuzijskoga liječenja i primjene čimbenika rasta granulocitnih kolonija. Bolesnici u toj fazi moraju biti u sterilnoj jedinici. Porast granulocita prvi je koji se primjećuje kod tih bolesnika, a očekuje se tijekom drugoga tjedna od transplantacije. Funkcija krvotvornoga sustava u smislu oporavka hematopoeze obično se oporavlja kroz dva mjeseca, dok je imunološki oporavak nešto sporiji i započinje od trećega mjeseca.
Komplikacije. Glavne komplikacije koje se povezuju s transplantacijom matičnih stanica su infekcije, reakcija presatka protiv primatelja (GvHD, engl. graft versus host disease) te rane i kasne toksične komplikacije intenzivne kemoradioterapije (mijeloablativno liječenje) koje su svojstvene i drugim oblicima onkološkoga liječenja.
Infekcije. U ranoj poslijetransplantacijskoj fazi dominira povećan rizik od nastanka bakterijskih infekcija i gljivičnih infekcija, dok u kasnoj fazi dominira rizik od nastanka virusnih infekcija i infekcije Pneumocystis carnii, kao i infekcijama uzrokovanim inkapsuliranim bakterijama (Streptococcus pneumoniae, Haemophylus influenzae, Neisseria meningitidis).
Reakcija presatka protiv primatelja (GvHD). Radi se o imunološki posredovanoj reakciji citotoksičnih T-limfocita iz presatka protiv tkiva domaćina, a javlja se kada presadak sadrži imunokompetentne stanice, kada postoji određena HLA-nepodudarnost davatelja i primatelja te kada je primatelj imunosuprimiran. Taj se poremećaj može prezentirati u dva oblika: akutni i kronični. Akutni oblik se javlja unutar stotinu dana od transplantacije, a prezentira se kožnim promjenama te zahvaćanjem jetre i organa probavne cijevi. Kronični se oblik nadovezuje na akutni ili se javlja de novo. U najtežim je oblicima moguć razvoj višestrukoga organskog zatajenja. Liječenje se provodi kortikosteroidima i imunosupresivima.
HITNA STANJA U HEMATOLOGIJI
Najčešća su hitna stanja u hematologiji febrilna neutropenija i neutropenična sepsa, hiperleukocitoza i sindrom leukostaze te sindrom lize tumora.
FEBRILNA NEUTROPENIJA
Definicija. Febrilna neutropenija česta je, po život opasna komplikacija, koja se susreće kod hematološko-onkoloških bolesnika, a definira se povišenom tjelesnom temperaturom iznad 38,5 ℃ u trajanju duljem od jednoga sata uz apsolutni broj neutrofila manji od 0,5 x 109/L ili manji od 1 x 109/L, ako se očekuje njihov daljnji pad na 0,5 x 109/L u sljedećih 24 do 48 sati (apsolutni broj neutrofila računa se tako da se postotak neutrofila u diferencijalnoj krvnoj slici podjeli sa 100 i pomnoži s ukupnim brojem leukocita). Bolesnici s febrilnom neutropenijom imaju visok rizik od nastanku bakterijskih i gljivičnih infekcija te svako odgađanje početka liječenja povećava mortalitet i smanjuje šansu za oporavak.
Etiopatogeneza. Febrilna neutropenija najčešće nastaje kao posljedica citotoksičnoga učinka kemoterapeutika na progenitorne stanice koštane srži gdje uzrokuju oštećenje molekule DNA, što rezultira smanjenim stvaranjem krvnih stanica. Zbog toga su osim neutropenije kod takvih bolesnika obično prisutne i anemija i trombocitopenija (pancitopenija). Osim toga, ona se može javiti i kao posljedica radioterapije ako se zračenjem obuhvaća velik dio aktivne koštane srži, ili kao posljedica tumorske infiltracije koštane srži. Kao posljedica manjka neutrofila i nezadovoljavajućega odgovora imunološkoga sustava bolesnici su skloni razvoju bakterijskih i gljivičnih infekcija i sepse (tzv. neutropenična sepsa). Glavni čimbenici rizika od razvoja infekcija kod takvih su bolesnika apsolutni broj neutrofila manji od 1,5 x 109/L, trajanje neutropenije dulje od sedam dana te bolesnikove teško opće stanje i komorbiditeti.
Mikrobiologija. Kod bolesnika s febrilnom neutropenijom najčešće se razvijaju gram-negativne bakterijske infekcije (Escherichia coli, Pseudomanas aeruginosa, Klebsiella pneumoniae), ali su sve češće i gram-pozitivne infekcije (stafilokokne i streptokokne infekcije), kao i gljivične infekcije (Candida spp., Aspergillus spp.). Također, bolesnici imaju povećan rizik i od nastanka virusnih infekcija (influenza, CMV, EBV).
Klinička slika. Osnovna klinička manifestacija febrilne neutropenije povišena je tjelesna temperatura, no treba imati na umu da ona može izostati kod bolesnika starije životne dobi, anergičnih bolesnika i bolesnika na kortikosteroidnoj terapiji. Uz povišenu tjelesnu temperaturu obično se nadovezuju i kliničke manifestacije razvijene infekcije i sepse, iako moraju biti razvijene kod svih bolesnika. Najčešća mjesta nastanka infekcija se kod takvih bolesnika: krv (neutropenična sepsa), respiratorni sustav (sinusitis, upala pluća, aspergiloza), probavni sustav (mukozitis, stomatitis, ezofagitis, gastroenteritis), koža (celulitis) te spolni sustav (vulvovaginitis, Fournierova gangrena).
Dijagnostička obrada. Dijagnoza febrilne neutropenije postavlja se na temelju laboratorijskih nalaza (apsolutni broj neutrofila) i prisutnosti povišene tjelesne temperature, ali sam dijagnostički postupak sastoji se od: 1) detaljne anamneze i fizikalnoga pregleda koji mogu upućivati na žarište infekcije, 2) osnovnih laboratorijskih nalaza (kompletna i diferencijalna krvna slika, biokemijske pretrage krvi i urina, koagulogram), 3) radioloških snimaka pluća (obično klasični radiografski snimak pluća, iako je dokazano da se kod više od polovice bolesnika kod kojih je klasični rendgenogram bio uredan na temelju nalaza CT-a mogu dokazati upalni infiltrati), 4) ultrazvuka abdomena (upalno žarište, limfadenopatija, splenomegalija) te 5) mikrobiološkoga uzorkovanja (hemokulture s minimalno dva odvojena mjesta, urinokultura, kultura septuma, koprokultura, brisevi sumnjivih mjesta i sl.). Prema indikaciji se dijagnostička obrada može proširiti na druge pretrage, kao što su lumbalna punkcija, biopsija koštane srži, serološka i molekularna dijagnostika virusnih infekcija, radiološko snimanje sinusa i sl.
Procjena rizika. Kako bi se bolesnici stratificirali u grupe s niskim i visokim rizikom od razvoja infekcija, a to ima ključnu u određivanju terapijskoga pristupa, danas su razvijeni različiti bodovni sustavi, a jedan takav je i MASCC sustav (engl. Multinational association for supportive care in cancer) prikazan u Tablici 6.47.
|
Tablica 6.47. Procjena rizika od razvoja infekcije kod neutropeničnih bolesnika
|
|
Karakteristika
|
Broj bodova
|
|
Opseg (znaci) bolesti
|
|
|
Bez simptoma ili blagi simptomi
|
5
|
|
Umjereni simptomi
|
3
|
|
Jako izraženi simptomi
|
0
|
|
Bez hipotenzije (RR < 90 mmHg)
|
5
|
|
Odsutnost kronične opstruktivne plućne bolesti
|
4
|
|
Solidni tumor ili nepostojanje gljivične infekcije
|
4
|
|
Bez dehidracije
|
3
|
|
Pojava vrućice u izvanbolničkim uvjetima
|
3
|
|
Dob < 60 godina
|
2
|
|
Najviša bodovna vrijednost je 26, a zbroj bodova preko 21 označava bolesnika niskoga rizika s febrilnom neutropenijom (ima rizik za razvoj teške infekcije manji od 5 % i smrtnost manju od 1 %).
|
Liječenje bolesnika s febrilnom neutropenijom treba biti brzo i energično s obzirom na mogućnost nastanka teških, po život opasnih infekcija. Terapijski pristup koji u prvome redu podrazumijeva izbor i način primjene antibiotika ovisi o stratifikaciji bolesnika u jednu od dvije skupine: niskorizični i visokorizični bolesnici (po prethodno opisanome MASCC sustavu). U određenim okolnostima u liječenje treba uvesti i antimikotike i antivirotike. Nakon početne terapije (prije koje je obvezno provesti mikrobiološko uzorkovanje) prva se reevaluacija radi nakon tri do pet dana, a daljnji nastavak liječenja ovisi o prispijeću mikrobioloških nalaza i/ili kliničkome odgovoru. Liječenje se provodi u bolničkim ustanovama, po mogućnosti u izoliranim prostorima.
Inicijalno liječenje niskorizičnih bolesnika (MASCC ≥ 21). Peroralna primjena antibiotika kod niskorizičnih bolesnika rezervirana je samo za bolesnike koji su hemodinamski stabilni, koji nemaju upalu pluća ili teške infekcije mekih tkiva, koji nemaju dugotrajne venske katetere te nemaju akutnu leukemiju ili znakove popuštanja drugih organa. Kod njih se klasično primjenjuje ciprofloksacin zajedno s koamoksiklavom (amoksicilin + klavulonska kiselina). Za sve je ostale niskorizične bolesnike indicirana intravenska primjena antibiotika bilo kao monoterapija (cefepim, piperacilin/tazobakatam ili karbapenem), bilo kao kombinacijska terapija (aminoglikozid + cefepim, piperacilin/tazobakatam ili karbapenem).
Inicijalno liječenje visokorizičnih bolesnika (MASCC ≤ 20). Kod takvih je bolesnika liječenje strogo ograničeno na intravensku primjenu antibiotika, a glavnu dilemu predstavlja odluka o inicijalnom uvođenju vankomicina u terapiju. Inicijalna primjena vankomicina u liječenju indicirana je kod bolesnika s prisutnim rizičnim faktorima za gram-pozitivnu bakterijemiju, kao i u ustanovama koje imaju visoku stopu gram-pozitivnih bakterijemija. Kod bolesnika kojima vankomicin nije potreban liječenje se provodi intravenskom primjenom antibiotika, bilo u monoterapiji bilo u kombinacijskoj terapiji, kao što je to opisano kod niskorizičnih bolesnika, dok se kod bolesnika kod kojih je potreban vankomicin primjenjuje antibiotska terapija koju čine vankomicin + cefepim, piperacilin/tazobakatam ili karbapenem ± aminoglikozid.
Reevaluacija terapije. Prva se procjena učinka antibiotske terapije obavlja tri do pet dana od početka terapije, a daljnje postupanje ovisi o prispijeću mikrobioloških nalaza i kliničkom odgovoru bolesnika.
Ako je izoliran mikrobiološki patogen, liječenje treba prilagoditi nalazu antibiograma, a ako uzročnik nije identificiran, daljnje postupanje ovisi o kliničkom odgovoru bolesnika, tj. je li bolesnik postao afebrilan ili je stanje i dalje febrilno.
Ako je bolesnik postao afebrilan, a pripada skupini niskorizičnih bolesnika, može ga se otpustiti na kućno liječenje uz nastavak peroralnoga liječenja ciprofloksacinom i koamoksiklavom, dok se kod visoko rizičnih bolesnika nastavlja sa započetom antimikrobnom terapijom.
Ako je kod bolesnika i dalje prisutna vrućica, treba pomišljati na njezine moguće uzroke: 1) radi se o rezistentnom bakterijskom soju, 2) bakterijska infekcija povezana je s nekrozom tkiva, 3) uzročnik je bakterija bez staničnoga zida i mogućnosti izolacije ili 4) vrućicu uzrokuju nebakterijski patogeni (gljivice, virusi) ili druge okolnosti (maligno stanje, transfuzija). Kod stabilnih bolesnika koji i dalje imaju vrućicu nastavlja se s incijalnom terapijom, dok kod bolesnika kod kojih se prati dodatno pogoršanje treba u terapiju uvesti vankomicin (ako nije uveden) i antifugalnu terapiju. Kod takvih je bolesnika potrebno osnovno mikrobiološko uzorkovanje te reevaluacija kroz tri do pet dana.
Kod primjene vankomicina kao rezervnoga antibiotika treba imati na umu da se njegova primjena mora strogo ograničiti na bolesnike sa stvarnom indikacijom i da njegova primjena mora prestati nakon tri dana (bilo da je uključen kao inicijalni lijek bilo kao lijek drugoga izbora) ako nema kliničkoga poboljšanja ili nije dokazana gram-pozitivna bakterija, kako bi se spriječio razvoj rezistencije. Kod bolesnika s narušenom bubrežnom funkcijom, kao i kod bolesnika s upalom pluća, umjesto vankomicina može se primijeniti linezolid.
Duljina trajanja liječenja. Antibiotska terapija se prekida kada se zadovolje sljedeći kriteriji: apsolutni broj neutrofila prelazi 0,5 x 109/L kroz dva dana, nema kliničkih i mikrobioloških dokaza infekcije te je bolesnik afebrilan dulje od 48 sati ili je apsolutan broj neutrofila manji od 0,5 x 109/L kroz sedam dana, a radi se o niskorizičnim bolesnicima kojima se nije pogoršavalo stanje niti se razvile komplikacije te je bolesnik afebrilan kroz pet do sedam dana.
Ostale napomene. Antivirusni lijekovi ne primjenjuju se rutinski kod svih bolesnika, nego samo kada postoji jasna kliničko-laboratorijska. Također, primjena faktora stimulacije granulocitopeze (G-CSF) ne primjenjuje se kod svih bolesnika, nego samo kod onih kod kojih se može očekivati pogoršanje kliničkoga stanja ili je ono znatno narušeno na samome početku liječenja. Najčešće se u tu svrhu primjenjuje filgrastim u dozi od 0,5 mcg/kg/dan, subkutano (ili kao intravenska infuzija) svaki dan do oporavaka broja neutrofila (početni porast očekuje se kroz dva do tri dana, no do punoga je učinka nekada potrebno i više tjedana liječenja).
HIPERLEUKOCITOZA I SINDROM LEUKOSTAZE
Definicija. Hiperleukocitoza je naziv za laboratorijski nalaz karakteriziran izraženom leukocitozom, odnosno broj leukocita veći od 100 x 109/L, dok se sindrom leukostaze definira kao hiperleukocitoza uz pridružene kliničke manifestacije. Radi se o potencijalno smrtonosnome stanju pri kojemu se stopa mortaliteta kreće između 30 i 40 % u prvome tjednu liječenja.
Etiologija. Hiperleukocitozu i sindrom leukostaze susrećemo kod bolesnika s akutnim i kroničnim leukemijama. Moguće ga je otkriti kod 10 do 20 % bolesnika s novootkrivenom akutnom mijeloičnom leukemijom, posebno kod bolesnika s mijelomonocitnom (FAB-M4), monocitnom (FAB-M5) te promijelocitnom leukemijom (FAB-M3). Također, taj poremećaj nalazimo kod 10 do 30 % bolesnika s akutnom limfoblastičnom leukemijom, a češći je kod djece i mlađih osoba, osoba muškoga spola te kod oboljelih od leukemija T-staničnoga fenotipa. U kroničnoj limfatičnoj leukemiji hiperleukocitoza je česta pojava (leukociti znaju biti i viši od 400 x 109/L), ali se sindrom leukostaze rijetko viđa. Također, sindrom leukostaze rijedak je i kod bolesnika s kroničnom mijeloičnom leukemijom.
Patofiziologija. Posljedice hiperleukocitoze objašnjavaju se teorijom povećane viskoznosti krvi te teorijom citokinskoga oštećenja. Teorija povećane viskoznosti krvi podrazumijeva nastanak oštećenja tkiva i organa kao posljedice agregacije i formiranja čepova u mikrocirkulaciji (posebno kada se radi o blastima), što dovodi do posljedične hipoperfuzije i hipoksemije (ishemijsko oštećenje). Teorija citokinskoga oštećenja podrazumijeva povećanu produkciju citokina iz aktivnih tumorskih stanica (blasti), a oni izazivaju oštećenje i posljedičnu aktivaciju endotela i drugih stanica.
Klinička slika sindroma leukostaze uključuje opće manifestacije (gubitak teka, umor, mršavljenje, febrilno stanje), respiratorne manifestacije (dispneja), neurološke manifestacije (vrtoglavica, poremećaji vida, glavobolja, poremećaj stanja svijesti) te druga očitovanja (pogoršanje renalne funkcije, ishemija udova, prijapizam, crijevna ishemija, koronarna ishemija i sl.).
Dijagnoza se postavlja na osnovi temeljnoga laboratorijskog nalaza hiperleukocitoze udruženoga sa specifičnim kliničkim manifestacijama, a osobito ako se od ranije zna da bolesnik ima leukemiju. Pored leukocitoze se u laboratorijskim nalazima često nalaze i hipoksemija, hiperkapnija, hiperkalijemija, hiperfosfatemija, hiperkalcijemija, hiperuricemija, azotemija, hiperlaktatemija, metabolička acidoza te lažna trombocitoza (brojač trombocita može brojati i fragmente blasta kao trombocite).
Liječenje. Osnovni terapijski cilj je kod ovih bolesnika citoredukcija. Prvi izbor pri tome predstavlja indukcijska kemoterapija, a ako je ona kontraindicirana ili se ne može odmah provoditi, citoredukcija se ostvaruje primjenom hidroksiureje (kod bolesnika s hiperleukocitozom) ili primjenom hidroksiureje zajedno s postupkom leukafereze (kod bolesnika sa sindromom leukostaze).
Indukcijska kemoterapija ključna je komponenta uspješnoga liječenja, a smanjenje broja leukocita ostvaruje se već unutar 24 sata. Ona nosi povećan rizik od nastanka sindroma lize tumora. Izbor kemoterapijskoga protokola ovisi o vrsti leukemije.
Hidroksiurjea je indicirana za citoredukciju kod bolesnika s hiperleukocitozom kod kojih se ne kreće odmah s indukcijskom kemoterapijom. Dnevna doza od 50 - 100 mg/kg/dan dovodi do redukcije 50 – 80 % broja leukocita unutar 24 - 48 sati, a cilj je terapije smanjiti broj leukocita ispod 50 x 109/L. Glavne su nuspojave vrućica, sindrom lize tumora te abnormalni jetreni nalazi.
Leukafereza se provodi kod bolesnika sa sindromom leukostaze i kontraindiciranom indukcijskom kemoterapijom, i to zajedno s primjenom hidroksiureje. Postupak leukafereze podrazumijeva aktivno prikupljanje krvi iz bolesnika u separator u kojemu se odvajaju leukociti, a potom se pročišćena krv vraća nazad u bolesnika. Leukafereza je kontraindicirana kod bolesnika s akutnom promijelocitnom leukemijom zbog pogoršanja intrizične koagulopatije.
Simptomatsko liječenje podrazumijeva dostatnu hidraciju, primjenu alopurinola te korekciju elektrolitnih poremećaja. Kod takvih bolesnika treba izbjegavati transfuzije eritrocita i prekomjernu primjenu diuretika s obzirom na to da dodatno povećavaju viskoznost krvi.
Prognoza. Loši ishodi pozitivno koreliraju s težinom kliničke slike, ali ne i s brojem leukocita. Stopa smrtnosti bolesnika sa sindromom leukostaze kreće se i do 40 %.
SINDROM LIZE TUMORA
Sindrom lize tumora nastaje kao posljedica kemoterapijskoga liječenja, a obilježen je destrukcijom velikoga broja visokoproliferativnih stanica pri čemu njihovi raspadni produkti dospijevaju u krv i izazivaju brojne metaboličke promjene i zatajenje organa i organskih sustava. Glavne kliničke manifestacije toga sindroma su hiperuricemija, hiperkalijemija, hiperkalcijemija, hiperfosfatemija i metabolička acidoza s njihovim posljedičnim očitovanjima (oligurija, kongestivno srčano popuštanje, poremećaji stanja svijesti, srčane aritmije, tetanija, konvulzije i sl.). Simptomi i znakovi obično se pojavljuju unutar 24 do 48 sati od početka liječenja. Liječenje je simptomatsko (potporno), a ključ leži u odgovarajućoj prevenciji koja započinje prije same kemoterapije te traje tijekom i nakon nje, a uključuje zadovoljavajuću hidraciju, primjenu alopurinola (300 mg/dan) te alkalizaciju urina.
- Abrams C.S. Thrombocytopenia. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition., Philadelphia: Elsevier Saunders; 2016. p. 1159-67.
- Andrašek I, Ravlić M, Cmrečak F, Orešković D, Deveđija S i sur. Hitna stanja u onkologiji. Lib Oncol. 2019;47(2-3):103–7
- Annibale B, Lahner E, Fave GD. Diagnosis and management of pernicious anemia. Curr Gastroenterol Rep. 2011 Dec;13(6):518-24.
- Antony A.C. Megaloblastic Anemias. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1104-13.
- Appelbaum F.R. The Acute Leukemias. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1239-46.
- Auerbach M, Deloughery T. Single-dose intravenous iron for iron deficiency: a new paradigm. Hematology Am Soc Hematol Educ Program. 2016 Dec 2;2016(1):57-66.
- Aurer I. Escencijalna trombocitemija. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izd. Zagreb: Ljevak; 2008. str. 990-1.
- Aurer I. Indolentni limfomi. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izd. Zagreb: Ljevak; 2008. p. 1019-22.
- Aurer I. Policitemija rubra vera. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008. str. 986-8.
- Babarović E, Hadžisejdić I, Štifter S, Lučin K, Jonjić N. Utjecaj angiogeneze, izražaja osteopontina i vaskularnog endotelnog čimbenika rasta u koštanoj srži na odgovor na prvu liniju terapije u pacijenata s multiplim mijelomom. Medicina Fluminensis. 2016 Sep;52(3):390-9.
- Bacigalupo A, Giammarco S, Sica S. Bone marrow transplantation versus immunosuppressive therapy in patients with acquired severe aplastic anemia. Int J Hematol. 2016 Aug;104(2):168-74
- Bagby G.C. Aplastic Anemia and Related Bone Marrow Failure States. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 114-1121.
- Bain B.J. The peripheral blood smear. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1052-9.
- Barbui T, Thiele J, Gisslinger H, Kvasnicka HM, Vannucchi AM, Guglielmelli P, Orazi A, Tefferi A. The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: document summary and in-depth discussion. Blood Cancer J. 2018 Feb 9;8(2):15.
- Bašić-Kinda S, Dujmović, Radman I, Serventi–Seiwerth R. Aurer I. Liječenje bolesnika s uznapredovanim stadijem Hodgkinova limfoma eskaliranim BEACOPP-om. Medicina Fluminensis. 2011 Sep 11;47(4):420-425.
- Batinić D. Imunološke pretrage u hematologiji. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008. str. 925-27.
- Berliner N. Leukocytosis and Leukopenia. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1130-8.
- Bierman P.J, Armitage J.O. Approach to the Patient with Lymphadenopathy nad Splenomegaly. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1138-42.
- Bierman P.J, Armitage J.O. Non-Hodgkin Lymphomas. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1257-68.
- Bunn H.F. Approach to the anemias. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1059-68.
- Cappellini M.D. The Thalassemias. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1089-95.
- Chiappella A, Castellino A, Nicolosi M, Santambrogio E, Vitolo U. Diffuse Large B-cell Lymphoma in the elderly: standard treatment and new perspectives. Expert Rev Hematol. 2017 Apr;10(4):289-297
- Connors J.M. Hodgkin Lymphoma. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1268-73.
- Damon L.E., Andreadis C.B. Blood Disorders. Current Medical Diagnosis & Treatment. 58. Edition. New York: McGraw Hill Education; 2019. p. 5120-55.
- De Jong A, Eikenboom J. Developments in the diagnostic procedures for von Willebrand disease. J Thromb Haemost. 2016 Mar;14(3):449-60.
- Devalet B, Mullier F, Chatelain B, Dogné JM, Chatelain C. Pathophysiology, diagnosis, and treatment of paroxysmal nocturnal hemoglobinuria: a review. Eur J Haematol. 2015 Sep;95(3):190-8.
- Dingli D, Ailawadhi S, Bergsagel PL, Buadi FK, Dispenzieri A, Fonseca R et al. Therapy for relapsed multiple myeloma: guidelines from the Mayo stratification for myeloma and riskadapted therapy. Mayo Clin Proc. 2017 Apr;92(4):578–98.
- Dodig D, Ivančević D. Nuklearno-medicinske pretrage u hematologiji. U: Vrhovac B., Jakšić B. Reiner Ž., Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008., str. 929-31.
- Dominis M. Citološke i histološke pretrage. U: Vrhovac B., Jakšić B. Reiner Ž., Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008. str. 923-25.
- Dubravčić K., Batinić D. Imunofenotipizacija akutnih leukemija i minimalna ostatna bolest. Paediatr Croat. 2013; 57(1): 258-62.
- Duraković N, Radonić R, Gašparović V. Thrombotic Thrombocytopenic Purpura – The Role of ADAMTS13 Assay in Clinical Practice. Collegium antropologicum. 2010 Sep;34(3):1087-91.
- Farrell CJ, Kirsch SH, Herrmann M. Red cell or serum folate: what to do in clinical practice. Clin Chem Lab Med. 2013 Mar 1;51(3):555–69.
- Fraenkel PG. Understanding anemia of chronic disease. Hematology Am Soc Hematol Educ Program. 2015;2015:14–8.
- Gallagher P.G. Hemolytic Anemias: Red Blood Cell Membrane and Metabolic Defects. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1080-88.
- Gasper W.J, Rapp J.H, Jhonskon M.D. Blood Vassel and Lymphatic Disorders. Current Medical Diagnosis & Treatment. 58. Edition. New York: McGraw Hill Education; 2019. p. 483-509.
- Gavranić B.B, Bašić-Jukić N, Kes P. Razvoj heparinom inducirane trombocitopenije 24 godine nakon početka kronične dijalize. Acta medica Croatica. 2012; (66):68-71
- George LA, Sullivan SK, Giermasz A, Rasko JEJ, Samelson-Jones BJ, Ducore J et al. Hemophilia B gene therapy with a high-specific- activity factor IX variant. N Engl J Med. 2017 Dec 7; 377(23):2215–27.
- Ginder G.D. Microcytic and Hypochromic Anemia. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1068-73.
- Glogauer M. Disorders od Phagocyte Function. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1143-51.
- Go RS, Winters JL, Kay NE. How I treat autoimmune hemolytic anemia. Blood. 2017 Jun 1;129(22):2971-9.
- Gredelj Šimec Nj, Kaić G, Škrtić A, Šiftar Z, Lasan-Trčić R, Valković T i sur. Smjernice za dijagnozu i liječenje bolesnika s mijelodispastičnim sindromom. Liječ Vjesn, 2017;139:1–11
- Grković L., Labar B. Akutna mijeloična leukemija u odraslih: dijagnostika i liječenje. Medicina Fluminensis. 2011;47(4):335-342
- Gudbrandsdottir S, Birgens HS, Frederiksen H, Jensen BA, Jensen MK, Kjeldsen L et al. Rituximab and dexamethasone vs dexamethasone monotherapy in newly diagnosed patients with primary immune thrombocytopenia. Blood. 2013 Mar 14; 121(11):1976–81.
- Hallek M. Chronic lymphocytic leukemia: 2017 update on diagnosis, risk stratification, and treatment. Am J Hematol. 2017 Sep;92(9):946–65.
- Jagannath VA, Fedorowicz Z, Al Hajeri A, Sharma A. Hematopoietic stem cell transplantation for people with ß-thalassaemia major. Cochrane Database Syst Rev. 2016 Nov 30;11(11):CD008708
- Jakšić B, Pejša V, Ostojić-Kolonić S, Kardum-Skelin I, Bašić-Kinda S, Coha B i sur.. Smjernice za dijagnostiku i liječenje kronične limfocitne leukemije – Krohem B-CLL 2017. Acta clinica Croatica. 2018; 57(1):190-215
- Jakšić B. Limfoproliferativne bolesti. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008. str. 1003-12.
- Jakšić O., Pejša V. Struktura i funkcija imunopoetskog sustava. U: Vrhovac B., Jakšić B. Reiner Ž., Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008. str. 915-18.
- Kaić G, Jelić Puškarić B, Pažur M, Šunjić Stakor M, Ostojić Kolonić S, Radić Krišto D i sur. Mijelodisplastični sindrom – nove spoznaje i „stara“ morfologija. Liječnički vjesnik. 2020;141(7-8):209-13.
- Kaushansky K. Hematopoesis and Hematopoetic growth factors. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1050-2.
- Keating A., Bishop M.R. Hematopoetic Stem Cell Transplantation. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1198-204.
- Kennedy JA, Ebert BL. Clinical implications of genetic mutations in myelodysplastic syndrome. J Clin Oncol. 2017 Mar 20; 35(9):968–74. Rytting ME et al. Acute lymphoblastic leukemia in adolescents and young adults. Cancer. 2017 Jul 1;123(13):2398–403.
- Kessler CM, Knöbl P. Acquired haemophilia: an overview for clinical practice. European Journal of Haematology. 2015 Dec; 95(81):36–44.
- King A, Shenoy S. Evidence-based focused review of the status of hematopoietic stem cell transplantation as treatment of sickle cell disease and thalassemia. Blood. 2014 May 15; 123(20):3089–94.
- Kušec R. Idiopatska mijelofibroza. U: Vrhovac B, Jakšić B,. Reiner Ž., Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; str. 988-90.
- Kušec R. Sistemska mastocitoza. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008. str. 991-2.
- Kutleša V., Konja J., Topuzović N. Aplastična anemija nakon preboljelog virusnog hepatitisa. Med Vjesn 1998; 30(1-2): 93-6
- Labar B, Kušec R, Jakšić B, Škare-Libernjak Lj, Načinović-Duletić A, Petričević-sinčić J i sur. Dijagnostičko-terapijski pristup u bolesnika s esencijalnom trombocitopenijom. Liječnički Vjesnik 2010; 132:333–339.
- Labar B. Akutne leukemije. U: Vrhovac B., Jakšić B, Reiner Ž, Vucelić B, ur. Interna medicina. 4 . izdanje. Zagreb: Ljevak; 2008. str. 992-1001.
- Labar B. Dijagnostičke metode. U: Vrhovac B., Jakšić B, Reiner Ž., Vucelić B, urednici. Interna medicina. 4 . izdanje. Zagreb: Ljevak; 2008. str. 922-3.
- Labar B. Hereditarni poremećaji strukture i sinteze hemoglobina. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4 . izdanje. Zagreb: Ljevak; 2008. str. 946-7.
- Labar B. Stečeni i urođeni sindromi aplastične anemije. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4 . izdanje. Zagreb: Ljevak; 2008. str.947-51.
- Labar B. Struktura i funkcija koštane srži. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4 . izdanje. Zagreb: Ljevak; 2008. str. 913-5.
- Labar B. Transplntacija krvotvornih matičnih stanica. U: Vrhovac B., Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4 . izdanje. Zagreb: Ljevak; 2008.,str. 1037-9.
- Labar B., Jakšić B. Podjela bolesti krvotvornog sustava. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4 . izdanje. Zagreb: Ljevak; 2008., str. 920-2.
- Labar B., Ostojić S. Anemije zbog povećane ili ubrzane razgradnje eritrocita. U: Vrhovac B., Jakšić B, Reiner Ž., Vucelić B, urednici. Interna medicina. 4 . izdanje. Zagreb: Ljevak; 2008. str. 940-6.
- Lang N., Mingo H. Bolesti granulocita. U: Vrhovac B., Jakšić B, Reiner Ž., Vucelić B, urednici. Interna medicina. 4 . izdanje. Zagreb: Ljevak; 2008. str. 972-5
- Leavitt A.D., Minichiello T. Thrombosis and Antithrombotic Therapy. Current Medical Diagnosis & Treatment. 58. Edition. New York: McGraw Hill Education; 2019. p. 556-88.
- McKenzie SE, Sachais BS. Advances in the pathophysiology and treatment of heparin-induced thrombocytopenia. Curr Opin Hematol. 2014 Sep;21(5):380–7.
- Michel M. Autoimmune and Intravascular Hemolytic Anemias. . In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1073-80.
- Milunović V, Bogeljić Patekar M, Mandac Rogulj I, Planinc-Peraica A, Ostojić Kolonić S. Suvremeni pristup ne-Hodgkinovu limfomu plaštene zone: pregled literature. Liječnički vjesnik. 2016;(138)11-12
- Milunović V, Bogeljić Patekar M, Mišura Jakubac K, Rogulj Mandac I, Radić Krišto D, Ostojić Kolonić S i sur. Održavanje rituksimabom u uznapredovalom folikularnom limfomu: kontroverzije. Acta clinica Croatica, Acta clinica Croatica. 2017(1);143-56
- Milunović V, Mandac Rogulj I, Perica D, Mišura Jakobac K, Gredelj Šimec Nj, Škrtić A i sur. Prognostički čimbenici u mijelodisplastičnom sindromu: iskustvo jednog centra. Liječnički vjesnik . 2019;141(7-8):238-46
- Mrsić M. Bolesti monocita i makrofaga. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4 . izdanje. Zagreb: Ljevak; 2008., str. 975-6.
- Murković M, Valković T. Višestruki zloćudni tumori u pacijenta s leukemijom vlasastih stanica: prikaz slučaja. Medicina Fluminensis. 2018;54(4):433-7.
- Murphy M.F., Pasi K.J., Mead A. Haematological disease. In: Kumar P, Clark M. Kumar & Clark`s Medicine. 9. edition. London: Elsevier; 2017. p. 1264-423.
- Mužić K, Rački S. Anemija u kroničnoj bubrežnoj bolesti. Medicina Fluminensis.2010;46(4):471-81
- Nemet D. Anemija i druge manifestacije nedostatka željeza, vitamina B12 i folata. Medicus. 2000;9:59-71.
- Nemet D. Anemije u kroničnim bolestima i sistemnim poremećajima. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4 . izdanje. Zagreb: Ljevak; 2008., str. 951-6.
- Nemet D. Anemije zbog poremećaja metabolizma željeza. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4 . izdanje. Zagreb: Ljevak; 2008., str. 933-6.
- Nemet D. Klasifikacija i klinički znakovi anemije. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4 . izdanje. Zagreb: Ljevak; 2008. str. 931-2.
- Nemet D. Kronična mijeloična leukemija. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4 . izdanje. Zagreb: Ljevak; 2008., str. 982-6.
- Nemet D. Kultura krvotvornih matičnih stanica. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4 . izdanje. Zagreb: Ljevak; 2008., str. 924-5.
- Nemet D., Planinc-Peraica A. Neoplastičke bolesti plazma stanica. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4 . izdanje. Zagreb: Ljevak; 2008., str. 1026-36.
- Nichols W.L. Von Willenbrand Disease and Hemorrhagic Abnormalitates of Platelet and Vasczular Function. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1167-72.
- O'Brien S., Jabbour E. The Chronic Leukemias. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1246-57.
- Ostojić S., Jakšić B. Hitna stanja u onkologiji. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4 . izdanje. Zagreb: Ljevak; 2008. str. 1066-70.
- Pejša V., Mingo H. Hodgkinov limfom. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4 . izdanje. Zagreb: Ljevak; 2008. str. 1022-6
- Petranović D, Pilčič G, Duletić Načinović A, Radaković M. Febrilna neutropenija. Medicina Fluminensis. 2011;47(3):281-286
- Planinc-Peraica A, Jakšić B. Bolesti limfocita. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4 . izdanje. Zagreb: Ljevak; 2008. str. 976-8.
- Planinc-Peraica A, Jakšić B. Bolesti slezene. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4 . izdanje. Zagreb: Ljevak; 2008. str. 978-81.
- Planinc-Peraica A, Ostojić S. Maligni limfomi. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4 . izdanje. Zagreb: Ljevak; 2008. str. 1013-9.
- Planinc-Peraica A. Megaloblastične anemije. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4 . izdanje. Zagreb: Ljevak; 2008., str. 936-40.
- Radić Antolic M, Zadro R, Sertić D, Labar B. Praćenje odgovora bolesnika oboljelih od kronične mijeloične leukemije na liječenje inhibitorima tirozin-kinaze kvantitativnom lančanom reakcijom polimeraze u stvarnom vremenu. Biochemia Medica. 2009;19(1):63-72.
- Radman I, Vodanović M, Mandac-Rogulj I, Roganović J, Petranović D, Valković T i sur. Smjernice Hrvatskog društva za hematologiju HLZ-a i KROHEM-a za zbrinjavanje anemije uzrokovane manjkom željeza. Liječnički vjesnik. 2019;141(1-2):1-13
- Ragni M.V. Hemorrhagic Disorders: Coagulation factor Deficiencies. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. , Philadelphia: Elsevier Saunders; 2016. p. 1172-81.
- Rajkumar S.V. Plasma Cell Disorders. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1273-84.
- Ries S. Citomorfološka dijagnostika akutnih mijeloičnih leukemija – podjela prema Svjetskoj zdravstvenoj organizaciji. Medicina Fluminensis. 2011;47(4):353-359.
- Roganović J. Akutna limfoblastična leukemija u djece. Medicina Fluminensis . 2011;47(4):343-352
- Rothenberg M.E. Eosinophilic Syndromes. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1152-4.
- Rumi E , Cazzola M. Diagnosis, risk stratification, and response evaluation in classical myeloproliferative neoplasms. Blood. 2017 Feb 9; 129(6):680–92.
- Sambol K, Cikač T. Dijagnostički i terapijski pristup bolesniku s multiplim mijelomom - prikaz bolesnika. Medica Jadertina. 2018;48(4):257-267
- Sayani FA, Abrams CS. How I treat refractory thrombotic thrombocytopenic purpura. Blood. 2015 Jun 18;125(25):3860–7.
- Schafer A.I. Bleeding and Thrombosis. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1154-9.
- Schafer A.I. Hemorrhagic Disorders: Disseminates Intravascular Coagulation, Liver Failure and Vitamin K Deficiency. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1181-4.
- Schafer A.I. Thrombotic Disorders: Hypercoagulabile States. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. , Philadelphia: Elsevier Saunders; 2016. p. 1185-91.
- Steegmann JL, Baccarani M, Breccia M, Casado LF, García-Gutiérrez V, Hochhaus A et al. European LeukemiaNet recommendations for the management and avoidance of adverse events of treatment in chronic myeloid leukaemia. Leukemia. 2016 Aug;30(8): 1648–71.
- Steensma D.P., Stone R.M. Myelodisplastic Syndromes. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1233-9.
- Steinberg M.H. Sickle Cell Disease and Other Hemoglobinopathies. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1095-104.
- Stoof SC, Cnossen MH, de Maat MP, Leebeek FW, Kruip MJ. Side effects of desmopressin in patients with bleeding disorders. Haemophilia. 2016 Jan;22(1):39-45.
- Tefferi A. Polycithemia Vera, essential Thrombocythemia and Primary Myelofibrosis. In: Goldman L, Schafer A. Goldman-Cecil Medicine, 25th edition. Philadelphia: Elsevier Saunders; 2016. p. 1122-9.
- Tefferi A: Anemia in adults: A contemporary approach to diagnosis. Mayo Clin Proc 2003; 78:1274-1280.
- Tkalčić Švabek Ž, Josipović M, Franić Šimić I, Davidović-Mrsić S, Duraković N. Complex variant of Philadelphia translocation involving chromosomes 1, 9, 12 and 22 in a case with chronic myeloid leukemia (CML). Medicina Fluminensis. 2018;54(1):91-97
- Valentino LA. Blood-induced joint disease: the pathophysiology of hemophilic arthropathy. J Thromb Haemost. 2010 Sep;8(9):1895-902.
- Valković T. Esencijalna trombocitemija. Rad Hrvatske akademije znanosti i umjetnosti. Medicinske znanosti. 2015;(42):134.
- Vlašić Tanasković J. Laboratorijska dijagnstika monoklonalnih gamapatija. Glasnik pulske bolnice. 2014;11(11):14-9.
- Vrhovac R. Sindrom mijelodisplazije. In: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urrdnici. Interna medicina. 4. izdanje. Zagreb: Ljevak; 2008. str. 1001-3.
- Vukićević T. Akutna limfoblastna leukemija - dijagnostika i lečenje. Acta fac. med. 2002; 19(2):77-80.
- Watson H.G, Culligan D.J, Manson L.M. Haematology and Transfusion Medicine. In: Davidsons Principles and Practice of Medicine, 23rd Edition. London: Elsevier; 2018. p. 912-79.
- Wierda WG, Byrd JC, Abramson JS, Bhat S, Bociek G, Brander D et al. Hairy cell leukemia, version 2.2018, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2017 Nov;15(11):1414–27.
- Willrich MAV, Murray DL, Kyle RA. Laboratory testing for monoclonal gammopathies: focus on monoclonal gammopathy of undetermined significance and smoldering multiple myeloma. Clin Biochem. 2018 Jan;51:38–47.
- Zadro R., Davidović Mrsić S. Citogenetske i molekularne pretrage u hematologiji. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4 . izdanje. Zagreb: Ljevak; 2008. str. 927-9.
- Zupančić Šalek S. Bolesti uzrokovane poremećajima hemostaze. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4 . izdanje. Zagreb: Ljevak; 2008. str. 956-71.
- Zupančić-Šalek S, Pulanić D, Ostojić-Kolonić S, Pejša V, Valković T, Nemet D. Smjernice za dijagnostiku i liječenje primarne imunosne trmbocitopenije u odraslih. Liječ Vjesn 2017;139:192–198.
- Zupančić-Šalek S., Stančić V. Hemostaza. U: Vrhovac B, Jakšić B, Reiner Ž, Vucelić B, urednici. Interna medicina. 4 . izdanje. Zagreb: Ljevak; 2008. str. 918-20.
- Županić-Krmek D, Sučić M, Bekić D. Anemia Of Chronic Disease: Illness or Adaptive Mechanism. Acta clinica Croatica. 2014;53.(3.):348-353.